HOVON HO107 SCT
Main info
- Identifier:
- HO107 MOBILIZATION
- Sponsor:
- HOVON
- Working group party:
- SCT & Supportive care
- Age:
- 18-60
- Echelon:
- Level A
- Included patients:
-
38(of 60)
- Active sites:
-
4(of 4)
- Title:
The feasibility and efficacy of subcutaneous Plerixafor for mobilization of peripheral blood stem cells in allogeneic HLA–identical sibling donors: a prospective phase II study.
Timeline
News
22MAR2021: Close outs of all sites are finished and end of trial report is completed and submitted. Study is archived until 31Jan2034 (15 years after global end of trial).
21FEB2017: Study is closed for inclusion of new donors + patients because the primary endpoint has been reached. Follow Up phase continues until 2 years after registration of both donor and patient.
01JUL2016: Please note that the target of study subjects is 44. 30 donors+patients registered for subcutaneous arm (= current protocol) and 14 donors+patients were enrolled in the iv arm during the primary protocol phase (= before amendment of study design). Sites will be notified by email once the study is almost closed.
07SEP2015: Amendment of study design (closure of iv arm) is submitted and approved by Dutch EC. All sites are deactivated until required documents (LI sign. page protocol and local versions of ICF) are send to the HDC.
24NOV2014: After revision of the DSMB report no safety issues have arisen. The study is reopened.
14OCT2014: Following the advice of the DSMB, it is decided to temporarily close the study
23AUG2011: Please note: patient and donor need to sign a seperate informed consent.
The informed consent for a donor you can find under download documentation/forms -> General -> Patient Information & IC form (NL)
The informed consent for a patient you can find under download documentation/forms -> Other documents -> ho107 icf patient template v4 11may2011
Flow
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- Not any more
- Objectives:
Primary Objective:
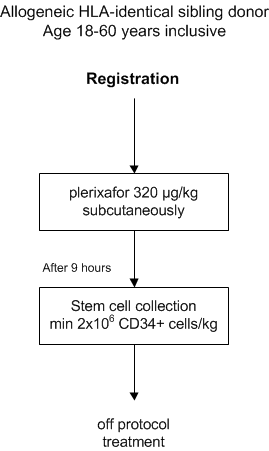
- To determine the feasibility of plerixafor 320 μg/kg subcutaneously to harvest a sufficient number of peripheral blood stem cells in healthy HLA-matched sibling donors.
Feasibility will be defined as follows: A harvest of at least 2x10^6 CD34+ cells/kg recipient body weight cells in one or two aphereses in at least 90% of the donors A harvest of at least 2x10^6 CD34+ cells/kg cells in one or two aphereses in 70% or less of the donors
will be considered as failure.Secondary Objectives:
Donors- To determine the efficacy of plerixafor subcutaneous. Efficacy will be determined as follows: The absolute number of CD34+ cells collected in 1 liter processed volume.
- To determine the time interval that is required to obtain 2.0x 106 CD34 + cells from start of mobilization procedure
- Pharmacokinetics: to determine the number of CD34+ cells in the peripheral blood at regular intervals after the administration of plerixafor.
- To determine the number of CD34+ cells in the peripheral blood as well as in the collected stem cells at regular intervals during the stem cell apheresis.
- To determine the phenotype of plerixafor mobilized and collected CD34+ cells including progenitor cells, dendritic cells and regulatory T-cells both in peripheral blood and collected stem cells.
- To document adverse events grade 1-4 during and directly following mobilization by plerixafor 320 μg/kg subcutaneously.
Patients
- To document engraftment 30, 60 and 90 days after transplantation with an allograft harvested after mobilization with plerixafor 320 μg/kg subcutaneous. Engraftment is defined as the first of 3 consecutive days with an absolute neutrophil count (ANC) of at least 0.5 ×10^9/l
- To document the day of hematopoietic reconstitution for neutrophils and platelets. This is defined as the first of 3 consecutive days of ANC ≥ 0.5x 10^6/L and platelets > 50x10^9/L
- To study chimerism in peripheral blood and T-cells 30, 60, and 90 days, and chimerism in bone marrow 90 days after transplantation with an allograft harvested after mobilization with plerixafor 320 μg/kg subcutaneously.
- To evaluate the incidence and grade of graft versus host disease (GVHD).
Eligibility
- Inclusion Criteria:
HLA identical sibling donor
- Age 18-60 years inclusive
- Hematologic parameters within normal limits
- Capable of undergoing leucapheresis: adequate venous access. Must be willing to undergo insertion of a central catheter should leucapheresis via peripheral vein be inadequate
- Willing and able to have bone marrow aspiration if there is mobilization failure
- Negative pregnancy test at study entry for women of childbearing potential
- Willing and able to use adequate contraception during the mobilization period and up to 3 months after last dose of plerixafor
- Written informed consent from donor
Patient inclusion criteria
- Age 18-65 years inclusive
- In general, indication for allogeneic stem cell transplant will be determined by each participating center according to local criteria.
- Patients with a cytopathologically confirmed diagnosis of:
- De novo Acute Myeloid Leukemia according to WHO classification in first complete remission (excluding acute promyelocytic leukemia)
- Therapy related AML/RAEB in first complete remission
- Myelodysplasia RA(RS)/RCMD with IPSS ≥ 1.5
- Myelodysplasia refractory anemia with excess of blasts (RAEB) with IPSS ≥ 1.5 in first complete remission
- Biphenotypic leukemia in first complete remission OR
- De novo B or T Lineage Acute Lymphatic Leukemia in first complete remission.
- Multiple myeloma, not included in other transplant study
- Hodgkin Lymphoma
- Non-Hodgkin lymphoma
- Chronic lymfocytic leukemia
- Chronic myeloid leukemia
- WHO performance score 0,1 or 2
- Patients should have an HLA- identical sibling donor
- Life expectancy >3 months
- Negative pregnancy test at study entry for women of childbearing potential
- Willing and able to use adequate contraception
- Written informed consent from patient
- Exclusion Criteria:
Donor exclusion criteria
- Monozygotic twin
- Unstable hypertension requiring more than 1 medication.
- Positive serology for hepatitis C or HbsAg
- Treatment with other investigational drugs
- HIV positivity
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
- Pregnant or breastfeeding female subject
Patient exclusion criteria
- Cardiac dysfunction as defined by:
- Myocardial infarction within the last 6 months of study entry
- Reduced left ventricular function with an ejection fraction < 50% as measured by MUGA scan or echocardiogram,
- Unstable angina
- Unstable cardiac arrhythmias
- Severe pulmonary dysfunction (CTCAE grade 3-4, see appendix B)
- Severe neurological or psychiatric disease
- Significant hepatic dysfunction (serum bilirubin ≥ 1.5 times upper limit of normal or transaminases ≥ 2.5 times upper limit of normal)
- Significant renal dysfunction (creatinine clearance < 50 ml/min after rehydration)
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, cancer, etc.)
- Patient known to be HIV-positive
- Pregnant or breast-feeding female patients.
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule
- Presence of other active non-hematological malignancy
Registration Details
Eligible donors and patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; Use goto eCRF button > select the [Patient tab] and click the [Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31.10.7041028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
For donors and patients the following information will be requested:
- Local donor/patient code (optional)
- Sex
- Date of birth
- Date written informed consent
- Specific items donor/patient gives consent for (see ICF)
- Eligibility criteria
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.