HOVON HO116 AML
Main info
- Identifier:
- HO116 AML
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- 18-70
- Stage:
- 1st Line
- Echelon:
- Level A
- Included patients:
-
140(of 60)
- Active sites:
-
7(of 10)
- Title:
A phase I/II feasibility study of the combination of panobinostat alone and panobinostat and decitabine prior to donor lymphocyte infusion in recipients of allogeneic stem cell transplantation with poor and very poor-risk AML
Timeline
News
02AUG2022: Global Last Patient Last Visit is reached. Patients were followed 5 years after registration. Last CRF data is currently collected and within 1 year after LPLV an end of trial report is created. Site close-outs will start in April 2023.
02AUG2017: As of today, 02 AUG 2017, the HOVON 116 AML trial named ‘A phase I/II feasibility study of panobinostat alone and the combination panobinostat and decitabine prior to donor lymphocyte infusion in recipients of allogeneic stem cell transplantation with poor and very poor-risk AML” is closed for inclusion of new patients.
In total 140 patients have been included.
All patients who have already been included in the HO 116 trial will finish their treatment according to protocol.
21SEP16: Amendment 03 approved by EC Netherlands. Please send in requested forms (site documents checklist, ICF + protocol signature page).
BE: all Belgium sites temporarily closed for inclusion of new patients until amendment has been approved in Belgium.
Current dose levels:
Patients 1-100: Panobinostat 20 mg/day on days 1, 4, 8 and 11 of each treatment cycle, and Decitabine 1 dd of 10 mg/m2 on day 1, 2 and 3 of each treatment cycle.
Patients 101-above: Panobinostat 20 mg/day on days 1, 4, 8 and 11 of each treatment cycle
Please note that currently both poor and very poor risk patients can be included.
26AUG2015: Based on the decision rules as defined in the protocol the dose level has been deescalated.
As from now, patients will be treated according to dose level 2reduced (see above).
Patients who have already started treatment on dose level 1 or 2 should continue in dose level 1 or 2.
Patients who are included at dose level 1 or 2 but did not start panobinostat/decitabine treatment yet, should start treatment at dose level 2reduced.
3JUL2015: Protocol version 7 has been approved in the Netherlands
30JAN2015: Based on the decision rules as defined in the protocol the dose level has been escalated.
As from now, patients will be treated according to dose level 2 (see above).
Patients who have already started treatment on dose level 1 should continue in dose level 1.
Patients who are included at dose level 1 but did not start panobinostat/decitabine treatment yet, should start treatment at dose level 2.
24MAR2014: Central approval for the HOVON 116 AML trial in Belgium has been obtained from the METC UZ Gent.
12NOV2013: Central approval for the HOVON 116 AML trial in The Netherlands has been obtained from the METC Erasmus MC Rotterdam.
Flow
Details
- Phase:
- Prospective Phase I/II study
- Monitoring Type:
- Not any more
- Objectives:
Primary objective
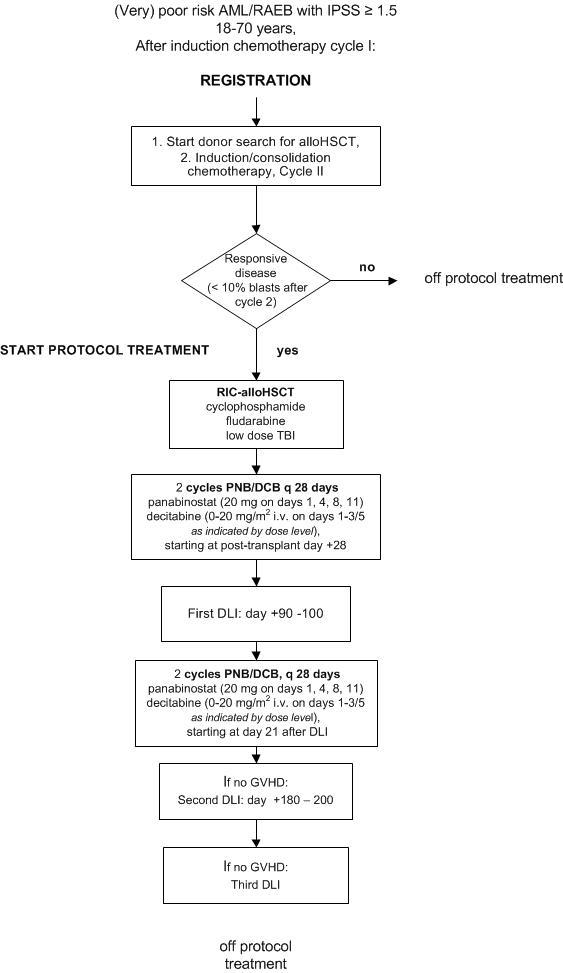
Part I- To assess the safety and feasibility of post-transplant panobinostat combined with decitabine to a regimen of T-cell replete RIC alloHSCT in patient with (very) poor-risk AML/RAEB, and select the recommended dose level for part II of the study
Part II
- To assess the feasibility and efficacy of addition of post-transplant panobinostat combined with decitabine to a regimen of T-cell replete RIC alloHSCT and DLI in patients with (very) poor-risk AML/RAEB
Part III
- To assess the feasibility and efficacy of Panobinostat monotherapy to a regimen of T-cell replete RIC alloHSCT and DLI in patients with (very) poor-risk AML/RAEB
Secondary objectives
To assess efficacy in terms of:- Complete remission rate at 3, 6, 12, and 24 months post alloHSCT
- Relapse/progression rate at 3, 6, 12, and 24 months post alloHSCT
- Overall survival (OS) from study registration as well as OS from start protocol treatment
- Progression free survival (PFS) from alloHSCT with relapse (for patients in CR) and progression (for patients in PR) as events
- Engraftment and chimerism at 3, 6, 12, and 24 months post alloHSCT
To assess toxicity in terms of:
- The incidence and nature of (serious) adverse events
- The incidence and severity of acute and chronic GvHD up to 24 m post alloHSCT
- NRM up to 24 months post-transplant
- Number and percentage of registered patients starting protocol treatment (eligible for alloHSCT)
- Number and percentage of patients receiving post-transplant epigenetic therapy after alloHSCT
- Number and percentage of patients receiving DLI after alloHSCT
Eligibility
- Inclusion Criteria:
- Poor-risk or very poor-risk AML or RAEB with IPSS ≥ 1.5.
- Responsive disease (< 10% blasts at 3 and/or 4 weeks after start of induction cycle II)
- Recovery of mucositis after preceding chemotherapy
- Absence of active opportunistic infections
- Absence of active CNS localisation
- HLA-compatible donor available (≥ 7/8 matched unrelated donor or fully matched sibling donor)
- WHO-performance status 0-2
- Written informed consent
- Exclusion Criteria:
- Severe cardiac dysfunction (NYHA classification III-IV, see appendix H)
- Severe pulmonary dysfunction (CTCAE grade III-IV, see appendix G)
- Severe neurological or psychiatric disease
- Significant hepatic dysfunction (serum bilirubin or transaminases ≥ 5 times upper limit of normal)
- Significant renal dysfunction (creatinine clearance < 30 ml/min after rehydration)
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, cancer, etc.)
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- Trial Online Process (TOP, https://www.hdc.hovon.nl/top). A logon to TOP can be requested at the HOVON Data Center for participants.
- By faxing the completed registration/randomization CRF +31.10.7041028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Local patient code (optional)
- Sex
- Date of birth
- Date written informed consent
- Date of diagnosis
- Cytogenetic risk classification
- Registration in other HOVON trial
- Planned date induction cycle II
- Specific items patient gives consent for (see ICF)
- Eligibility criteria
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.