HOVON HO134 Benign
Main info
- Identifier:
- HO134 MF
- Sponsor:
- HOVON
- Working group party:
- Benign hematology
- Age:
- 18-70
- Stage:
- 1st Line
- Echelon:
- Level A
- Included patients:
-
61(of 70)
- Active sites:
-
9(of 12)
- Title:
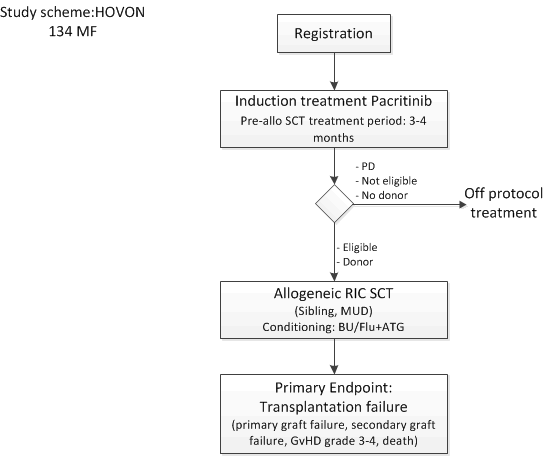
A phase II trial in patients with myelofibrosis (primary, post-ET or post PV-MF) treated with the selective JAK2 inhibitor pacritinib before reduced-intensity conditioning allogeneic stem cell transplantation.
Timeline
News
(No news)
Flow
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- Not any more
- Objectives:
Primary objective:
Improve allo-SCT transplant outcome using a uniform conditioning regimen and pacritinib pretreatment by means of the proportion of patients with a failure within 6 months post-transplant. Events that are considered a failure are: primary graft failure; secondary graft failure; acute GvHD grade 3-4; death whatever the cause is.Secondary objectives:
- To assess the effect of pacritinib treatment on splenomegaly
- To improve disease response by pacritinib treatment and allo-SCT as defined by the IWG-MRT response criteria
- To determine the proportion of patients receiving allo-SCT
- To document safety and toxicity of pacritinib treatment before allo-SCT
- To evaluate efficacy of induction therapy determined as time to response
- To determine time to red cell recovery (red blood cell transfusion incidence for >4 weeks) after SCT
- To determine progression free survival and overall survival from study inclusion and separately from allo-SCT in transplanted patients
- To evaluate NRM (Non-Relapse -Mortality) from inclusion and separately after allo-SCT in transplanted patients
- To determine effects on JAK2, calreticulin or mpl (allelic) burden before and after allo-SCT (allelic burden before pacritinib treatment, at conditioning and 1, 3 and 6 months post SCT)
- To summarize Quality-of-Life (QOL) during/after treatment using the MPN-SAF scoring tool
- To determine effects on inflammatory cytokines at study entry, before start conditioning and 3 and 6 months after allo-SCT
- To determine the prognostic value of additional molecular markers (ASXL1, TET2, SRSF2, DNMT3A, EZH2, CBL, IDH1,2, Lnk, SF3B1)
Eligibility
- Inclusion Criteria:
- Patients with a confirmed diagnosis of post-ET, post-PV or primary myelofibrosis
- Intermediate-2 or high-risk according to DIPSS plus
- Age 18-70 years inclusive
- WHO performance status 0-2
- Platelet count ≥ 25 × 10^9/L without platelet support within 2 weeks before study entry
- All men and women of childbearing potential must agree to use adequate contraception during the study
- Written informed consent
- Patient is capable of giving informed consent
- Exclusion Criteria:
- Patients who have been treated with pacritinib as their previous JAK2 inhibitor treatment cannot participate in this study
- Previous treatment with JAK2 inhbitors, other than pacritinib, is allowed with the exception of high dose ruxolitinib (above 10 mg BID). For these patients taper the dose to 10 mg BID or lower at least 2 weeks before pacritinib treatment is allowed
- Any GI or metabolic condition (e.g. inflammatory or chronic functional bowel disorder such as Crohn’s Disease, Inflammatory Bowel Disease, chronic diarrhea or constipation) that could interfere with absorption of oral medication
- Left ventricular cardiac ejection fraction of ≤ 45% by echocardiogram or multigated acquisition (MUGA) scan
- Impaired liver and renal function, defined by liver transaminases (aspartate aminotransferase [AST]/serum glutamic oxaloacetic transaminase [SGOT] and alanine aminotransferase [ALT]/serum glutamic pyruvic transaminase [SGPT]), >3 × the upper limit of normal (ULN) (AST/ALT >5 × ULN if transaminase elevation is related to MF), direct bilirubin >4× ULN, and creatinine clearance ˂ 40 ml/min.
- Impaired coagulation function, defined by prothrombin time (PT)/international normalized ratio (INR), partial thromboplastin time (APTT)>1.5 x ULN
- Experimental treatment within four weeks before inclusion for PMF, Post-PV, or Post-ET MF
- Severe pulmonary dysfunction (CTCAE grade III-IV, see appendix D)
- Treatment with a potent strong CYP3A4 inhibitor or a strong cytochrome P450 (CYP450) inducer within the last 2 weeks
- Treatment with anticoagulation or antiplatelet agents, except for aspirin dosages of ≤100 mg per day, within the last 2 weeks
- New York Heart Association Class II, III, or IV congestive heart failure
- QTc prolongation >450 ms as assessed by ECG and corrected by Federicia method or other factors that increase the risk for QT interval prolongation (e.g., heart failure, hypokalemia [defined as serum potassium <3.0 mEq/L that is persistent and refractory to correction], family history of long QT interval syndrome, or concomitant use of medications that may prolong QT interval)
- Significant recent bleeding history defined as NCI CTCAE grade ≥2 within the last 3 months, unless precipitated by an inciting event (e.g., surgery, trauma, injury)
- Any history of CTCAE grade ≥2 non-dysrhythmia cardiac conditions within the last 6 months. Patients with asymptomatic grade 2 non-dysrhythmia cardiac conditions may be considered for inclusion, with the approval of the principal investigator, if stable and unlikely to affect patient safety.
- Any history of CTCAE grade 2 cardiac dysrhythmias within the last 6 months. Patients with non-QTc CTCAE grade 2 cardiac dysrhythmias may be considered for inclusion, with the approval of the principal investigator, if the dysrhythmias are stable, asymptomatic, and unlikely to affect patient safety.
- Patients with active, uncontrolled infections
- Patients known to be HIV (human immunodeficiency virus)-positive
- Active hepatitis A, B or C
- History of active malignancy during the past 3 years, except basal carcinoma of the skin or stage 0 cervical carcinoma
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, cancer, etc.)
- Pregnant or breastfeeding women
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- ALEA (https://aleaclinical.com/Hovon/DM/DELogin.aspx?refererPath=DEHome.aspx). A logon to ALEA can be requested at the HOVON Data Center for participants
- By faxing the completed registration/randomization CRF +31.10.7041028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
HOVON interim/final analyses for the coming 6 months: Interim analysis of first transplanted patients (EHA-2020; Q1-2020)
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.