HOVON HO136 NHL
Main info
- Identifier:
- HO136 NHL
- Sponsor:
- HOVON
- Working group party:
- Lymphoma
- Age:
- >= 18
- Stage:
- 2nd Line
- Echelon:
- Level B
- Included patients:
-
36(of 37)
- Active sites:
-
16(of 17)
- Title:
Phase I-II study combining Brentuximab Vedotin with second line salvage chemotherapy (RDHAP) in CD30 positive diffuse large B-cell lymphoma patients refractory to first line chemotherapy or in first relapse who are eligible for high dose treatment followed by autologous stem cell transplantation.
Timeline
News
New SAE form available
01JUL2021:
The SAE form has been updated. The biosimilar Ruxience has been added as an option.
Continuation with phase II part of HOVON 136 NHL
31-07-2019: The safety analysis of the Phase I part of the HOVON 136 NHL study has been performed. The results indicate that the starting dose Level L is feasible.
This means the study will be reopened and that from now on patients can be included in the Phase II part of the study. Patients will be registered in TOP.
The current drug dose level is:
- Brentuximab vedotin 1.8 mg/kg
- Cisplatin 100 mg/m2
- Cytarabine 2 g/m2 q 12 hr
The Phase II registration CRF can be found on the HOVON-website
Registration of new patients on hold
21-03-2019: Currently, six patients are included in dose level L of the study. This means that the inclusion of new patients is on hold until the feasibility of the current dose level is determined by a safety analysis. Based on this, it is decided whether the phase 2 part can be continued.
Depending on the final number of Serious Toxicities that will be reported, the study will continue with:
- phase I, dose level L-1 (when ST has been reported in two or more patients)
- phase II (when the current dose level appears feasible, ST in one or less patients)
You will be informed about this (by mail and via the HOVON website) as soon as it is determined how the study will be continued.
Flow
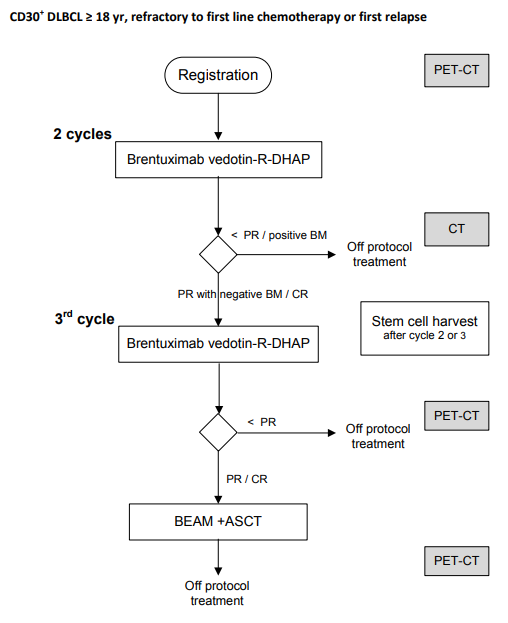
Details
- Phase:
- Prospective Phase I/II study
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
Phase I:
Primary objective- To identify the feasibility and RDL (recommended dose level) of brentuximab vedotin in combination with R-DHAP
Secondary objectives
- To assess the toxicity of brentuximab vedotin in combination with R-DHAP
- To assess the success rate of autologous peripheral blood stem cell harvest after brentuximab vedotin-R-DHAP
Phase II
Primary objectives- To evaluate the efficacy of the combination of brentuximab vedotin and R-DHAP as salvage treatment in relapse/refractory DLBCL patients in terms of metabolic CR rate after the third cycle
- To establish the rate of CTCAE grade 3/4 non-hematological toxicity, including neurotoxicity after each cycle of brentuximab vedotin-R-DHAP
Secondary objectives:
- To asses the overall response rate (ORR) after 3 cycles and after ASCT
- To assess the toxicity profile of brentuximab vedotin in combination with R-DHAP
- To assess hematological recovery after each cycle of brentuximab vedotin-R-DHAP
- To assess the success rate of harvesting an autologous peripheral blood stem cell graft
- To assess the fraction of patients (CR/PR) eligible for ASCT who actually undergo ASCT
- To assess peripheral blood neutrophil and platelet recovery after ASCT
- To evaluate the progression free survival (PFS), event free survival (EFS), and overall survival (OS)
- To identify predictive factors for response, PFS, EFS and OS (exploratory analysis)
Eligibility
- Inclusion Criteria:
- CD30 positive DLBCL, i.e. more than 1% of DLBCL cells CD30 positive (central pathology review results not required to enter patient into the study), according to the WHO classification 2008:
- CD30 positive DLBCL, including EBV positive DLBCL,
- CD30 positive primary mediastinal B-cell lymphoma.
- Primary refractory to or in first relapse after first line therapy with R-CHOP or R-CHOP-like therapy (A rituximab biosimilar is permitted when it is registered for the indication of DLBCL).
- Relapse is defined as biopsy confirmed CD30 positive DLBCL after a complete response. The relapse must be histologically confirmed. In case a surgical biopsy is not possible, at least confirmation by FNA biopsy is required.
- Refractory disease is defined as:
- 1) progressive disease during first line therapy, In this case biopsy confirmation of CD30 positive DLBCL is preferred but not required
- 2) stable disease after at least 3 cycles of first line therapy, In this case biopsy confirmation of CD30 positive DLBCL is preferred but not required.
- 3) PR after at least 6 cycles of first line therapy, or in the case of stage I-II disease after at least 3 cycles of therapy and definitive involved field radiotherapy. In this case refractory disease must be histologically confirmed.
- Age ≥ 18 years (upper age limit for ASCT at the discretion of the participating center).
- Measurable disease: on CT scan at least 1 lesion/node with a long axis of > 1.5 cm and at least one positive lesion on 18F-FDG PET scan.
- WHO performance status 0-2 (see appendix C).
- Adequate hepatic function: total bilirubin ≤ 1.5 times ULN (unless due to lymphoma involvement of the liver or a known history of Gilbert's syndrome as defined by > 80% unconjugated bilirubin) and ALAT/ASAT ≤ 3 times ULN (unless due to lymphoma involvement of the liver; in that case ALAT/ASAT may be elevated up to 5 times ULN).
- Adequate renal function: GFR > 60 ml/min as estimated by the Cockroft&Gault formula at rehydration: CrCL = (140-age [in years] x weight [kg] (x 0.85 for females) / (0.815 x serum creatinine [μmol/L]).
- Adequate bone marrow function: Absolute neutrophil count (ANC) ≥ 1.5x109/L and platelet count ≥ 100 x 109/L.
- Hemoglobin must be ≥ 8 g/dL (5.0 mmol/L), transfusion is allowed.
- Eligible for high-dose chemotherapy and ASCT.
- Resolution of relevant toxicities from first-line therapy.
- Life expectancy of > 3 months with treatment.
- Negative pregnancy test at study entry, if applicable.
- Female patient is either post-menopausal for at least 1 year before screening visit or surgically sterile or if of childbearing potential, agrees to practice 2 effective methods of contraception, at the same time, or agrees to completely abstain from heterosexual intercourse, from the time of signing the informed consent through 12 months after the last dose of study drug.
- Male patients, even if surgically sterilized, (i.e. status post vasectomy) agree to practice effective barrier contraception, or agrees to completely abstain from heterosexual intercourse, during the entire study period and through 12 months after the last dose of study drug.
- Written informed consent.
- Patient is capable of giving informed consent.
- CD30 positive DLBCL, i.e. more than 1% of DLBCL cells CD30 positive (central pathology review results not required to enter patient into the study), according to the WHO classification 2008:
- Exclusion Criteria:
- Peripheral sensory or motor neuropathy grade ≥ 2.
- Known cerebral or meningeal disease (NHL or any other etiology), including signs and symptoms of progressive multifocal leukoencephalopathy (PML).
- Symptomatic neurological disease compromising normal activities of daily living or requiring medications.
- Transformed lymphoma.
- DLBCL after organ transplantation.
- Immunodeficiency-associated B-cell lymphoproliferative disease.
- Use of other investigational agents within at least 5 half-lives of the most recent agent used prior to study entry.
- Treatment with myelosuppressive chemotherapy or biological therapy ≤ 4 weeks before study entry.
- Female patients who are breast feeding.
- History of another malignancy less than 3 years before study inclusion, or previously diagnosed with another malignancy and have evidence of residual disease, with the exception of non-melanoma skin cancer, completely resected melanoma TNMpT1 and carcinoma in situ of the uterine cervix.
- Known hypersensitivity to recombinant proteins, murine proteins, or to any excipient contained in the drug formulation of brentuximab vedotin.
- Active hepatitis B or C infection as defined by positive serology and transaminitis. Non-active hepatitis B carriers or anti-HBc positive patients may be included if protected with lamuvidine or entecavir (see 9.4).
- HIV positivity.
- Radiation therapy within 8 weeks prior to start of protocol treatment. Emergency radiation therapy is allowed, as long as measurable disease (at non-irradiated sites) persists.
- Patients with a serious psychiatric disorder that could, in the investigator's opinion, potentially interfere with the completion of treatment according to protocol.
- Major organ dysfunction, unless NHL-related.
- Patients who have any severe and/or uncontrolled medical condition or other conditions that could affect their participation in the study such as:
- Known history of symptomatic congestive heart failure (NYHA III, IV, appendix E), myocardial infarction ≤ 6 months prior to first study drug,
- Evidence of current serious uncontrolled cardiac arrhythmia, angina pectoris, electrocardiographic evidence of acute ischemia or active conduction system abnormalities,
- Recent evidence (within 6 months before first dose of study drug) of a left-ventricular ejection fraction <45%,
- Severely impaired pulmonary function as defined as spirometry and DLCO (diffusing capacity of the lung for carbon monoxide) that is 50% or less of the normal predicted value and/or O2 saturation that is 90% or less at rest on room air.
- Thyroid abnormalities when thyroid function cannot be maintained in the normal range by medication.
- Current participation in another clinical trial interfering with this trial.
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
- Claustrophobia to the extent that PET-CT is impossible.
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; Use goto eCRF button > select the [Patient tab] and click the [Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31 (0)10 704 1028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31 (0)10 704 1560 Monday through Friday, from 09:00 to 17:00 CET
Patients for the phase I part of the trial can only be registered by phone or by fax.
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Local patient code (optional)
- Sex
- (Partial) date of birth and age at time of registration
- Date written informed consent
- Specific items patient gives consent for (see ICF)
- Pathology number and original pathology laboratory
- Eligibility criteria
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.