TU Dresden HO138 AML
Main info
- Identifier:
- HOVON 138 AML
- Sponsor:
- TU Dresden
- Working group party:
- Leukemia
- Age:
- 18-65
- Stage:
- 1st Line
- Echelon:
- Limited Site Selection
- Included patients:
-
132(of 280)
- Active sites:
-
14(of 18)
- Title:
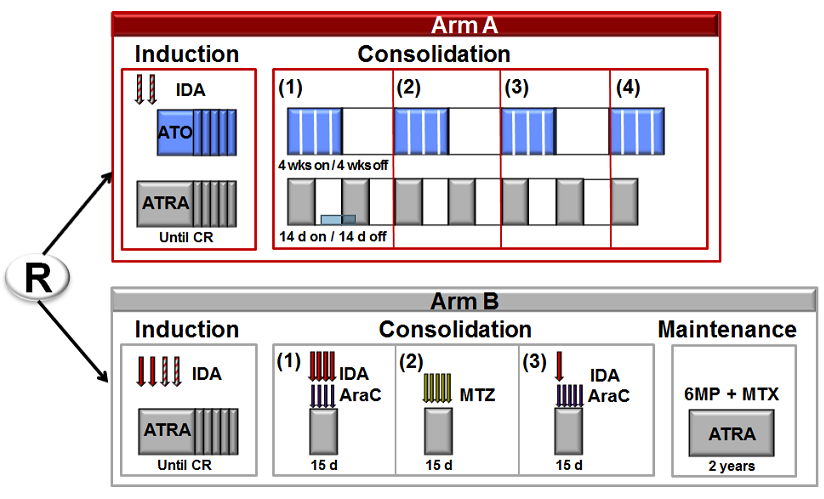
A randomized Phase III study to compare arsenic trioxide (ATO) combined to ATRA and idarubicin versus standard ATRA and anthracycLinces-based chemotherapy (AIDA regimen) for patients with newLy diagnosed high-risk acute prOmyelocytic leukemia
Timeline
News
13JAN2025: The study will be closed in all participating countries on January 21, 2025. After January 21, 2025, only the data collected up to that point will be evaluated and analyzed. Therefore, no transfer to CTIS will take place.
Flow
Details
- Phase:
- Prospective randomized Phase III study
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
The broad goal of QoL evaluation in this phase III study is to examine possible differences in keyQoL aspects of high-risk APL adult patients treated with ATRA-chemotherapy (standard arm) versus ATRA-ATO therapy (experimental arm). This information will help better understand the overall treatment effectiveness of the newer therapy (i.e., ATRA-ATO) being tested. Based on our previous report, key selected scales have been chosen for the primary objective. QoL results of this trial will be reported in accordance with high methodological quality criteria for documenting patient-reported outcomes in RCTs so recommended by the CONSORT PRO Extension.
Primary Objective
To investigate possible differences in the following a priori selected scales: physical and cognitive functioning as well as fatigue, nausea and vomiting, constipation and appetite loss (outcome measure: EORTC QLQ-C30).Secondary Objectives
- To investigate possible differences between treatment arms over time in all other QoL parameters assessed with the EORTC QLQ-C30 not considered in the primary objective.
- to evaluate the relationship between self-reported fatigue (outcome measure: EORTC QLQ-C30) FACIT-Fatigue scale) and the overall change over time in other QoL parameters measured by the EORTC QLQ-C30 (i.e., functional and other symptom aspects).
- To investigate the prognostic value of QoL parameters for overall survival. Fatigue and physical functioning will be regarded as the primary outcomes for prognostic factor analysis.
Eligibility
- Inclusion Criteria:
- Informed consent
- Women or men with a newly diagnosed APL by cytomorphology, confirmed by molecular analysis: The confirmation of diagnosis at genetic level (microspeckled PML nuclear distribution by PGM3 monoclonal antibody and/or PML/RARa fusion by RT-PCR or fluorescence in situ hybridization (FISH) and/or demonstration of t(15;17) at karyotyping) will be mandatory for patient eligibility. However, in order to avoid delay in treatment initiation, patients can be randomized on the basis of morphologic diagnosis only and before the results of genetic tests are available.
- Age ≥18 and ≤ 65 years
- ECOG performance status 0-3
- WBC at diagnosis > 10 GPt/l
- Serum total bilirubin ≤ 3.0 mg/dl (≤ 51 μmol/l)
- Serum creatinine ≤ 3.0 mg/dl (≤ 260 μmol/l)
- Women must fulfill at least one of the following criteria in order to be eligible for trial inclusion:
- Post-menopausal (12 months of natural amenorrhea or 6 months of amenorrhea with Serum FSH > 40 U/ml)
- Postoperative (i.e. 6 weeks) after bilateral ovariectomy with or without hysterectomy
- Continuous and correct application of a contraception method with a Pearl Index of <1% (e.g. implants, depots, oral contraceptives, intrauterine device– IUD).
- Sexual abstinence
- Vasectomy of the sexual partner
- Exclusion Criteria:
- Patients who are not eligible for chemotherapy as per discretion of the treating physician
- APL secondary to previous radio- or chemotherapy for non-APL disease
- Other active malignancy at time of study entry (exception: basal-cell carcinoma)
- Lack of diagnostic confirmation of APL at genetic level
- Significant arrhythmias, ECG abnormalities:
- Congenital long QT syndrome;
- History or presence of significant ventricular or atrial tachyarrhythmia;
- Clinically significant resting bradycardia (<50 beats per minute)
- QTc >500msec on screening ECG for both genders (using the QTcF formula detailed on protocol 5.5.6)
- Right bundle branch block plus left anterior hemiblock, bifascicular block
- Other cardiac contraindications for intensive chemotherapy (L-VEF <50%)
- Uncontrolled, life-threatening infections
- Severe non controlled pulmonary or cardiac disease
- Severe hepatic or renal dysfunction
- HIV and/or active hepatitis C infection
- Active multiple sclerosis (patients with inactive MS can be included)
- Pregnant or breast-feeding patients
- Allergy to trial medication or excipients in study medication
- Substance abuse; medical, psychological or social conditions that may interfere with the patients participation in the study or evaluation of the study results
- Use of other investigational drugs at the time of enrolment or within 30 days before study entry
Registration Details
The SAL study office provides an Investigator Site File to the study center. The ISF includes among others two randomization envelopes for the first patient of each participating study center. The randomization envelope includes among others the randomization result and the contact details for the online data base activation. This procedure guarantees a fast treatment start of the first patient. The second and following eligible patients of each participating center will be randomized by using the online data base. The data base of the SAL study office will automatically perform the randomization as described in chapter 11.2 and report the randomization result and the corresponding full patient ID number to the trial site and to the SAL study office via automatic email response. The full patient ID number will be noted in the patient chart and in the trial documentation forms. The randomization result will not be blinded, i.e. investigators will know whether patients will receive treatment A or treatment B.
15 May2020 interim analysis (DSMB)
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.