HOVON HO141 CLL
Main info
- Identifier:
- HO141 CLL
- Sponsor:
- HOVON
- Working group party:
- CLL
- Age:
- >= 18
- Stage:
- 2nd Line
- Included patients:
-
230(of 230)
- Active sites:
-
47(of 56)
- Title:
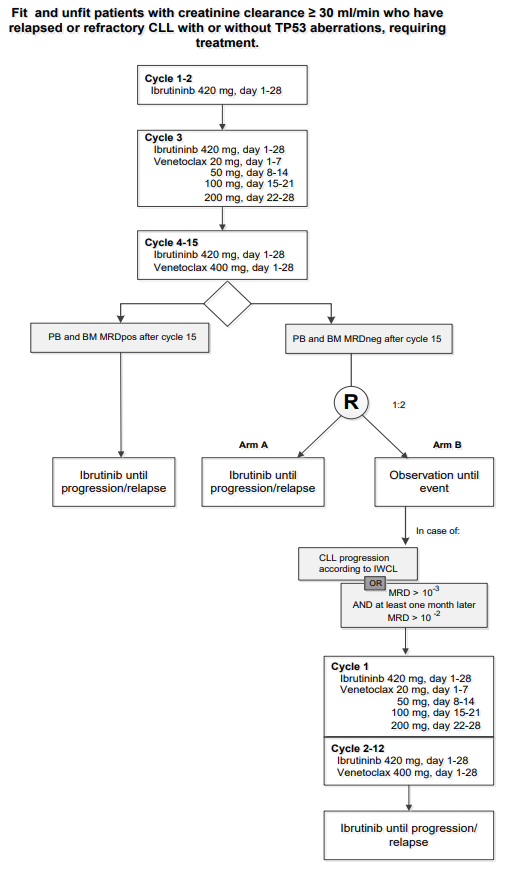
A prospective, multicenter, phase-II trial of ibrutinib plus venetoclax in patients with creatinine clearance ≥ 30 ml/min who have relapsed or refractory chronic lymphocytic leukemia (RR-CLL) with or without TP53 aberrations.
Timeline
News
20-Apr-2020: The updated pharmacy info letter V4 has been uploaded and can be found on the HOVON website.
14-Oct-2019: For Dutch sites only, a new biobank protocol and ICF has been implemented. The documents have been sent out to sites and can be found on the HOVON website. Further, tubes for additional blood samples have been sent to sites.
20-Mar-2019: Amendment 02 has been approved in the Netherlands.
The most important change is that the criterion for randomization after cycle 15 has been changed from ‘MRD negativity after cycle 12 (PB) AND at day 15 cycle 15 (PB and BM)’ to ‘MRD negativity at day 15 of cycle 15 (PB and BM)’. The randomization form has not been updated.
New documents:
*HO141_protocol-V04_20DEC201823-JAN-2019: The study is closed for inclusion of new patients.
08-NOV-2018: Amendment 01 has been approved.
New documents:
- HO141_protocol_AM01_V3_25JUL2018
- HO141_ICF_NL_v3_18JUL2018
- HO141_REGRAND_17OCT2018
- HO141 Pharmacy info letter_01NOV2018
- 20181009_HOVON141 Lab manual_1.8 - HOVON Sites –Final
20-JUL-2017: The study has been approved by EC, CA and the Biobank Committee in the Netherlands. All sites have been informed and may start with the local submission.
10-JUL-2017: The study has been approved in Denmark and the first site will open shortly.
The Netherlands is still awaiting approval from the Biobank Committee. As soon as we receive approval, the ITF will be sent to the sites.
Flow
Details
- Phase:
- Prospective randomized Phase II study
- Monitoring Type:
- Not any more
- Objectives:
The primary objective:
- Evaluate efficacy of ibrutinib + venetoclax (VI) in terms of proportion of patients fulfilling the criteria for progression free survival (PFS) at 12 months after stopping therapy (27 months after starting treatment) for patients randomized to stop treatment (arm B of the study), reinitiated treatment due to MRD positivity not considered progression (see section 13.1 for details).
Secondary objectives (for all treatment groups):
- Evaluation of the efficacy in terms of minimal residual disease (MRD) at 12 months after stopping treatment (month 27),
- Evaluation of efficacy in terms of PFS (IWCLL criteria),
- Time to next CLL treatment,
- MRD after cycle 12 (PB), day 15 cycle of 15 (PB and BM) and at later time points in PB,
- Overall Survival (OS),
- Complete response (CR)/Partial response (PR)/Stable disease (SD) after cycle 3, 9, 12, 15, and at month 27 and month 51 (3 years after stopping treatment),
- Duration of response,
- To evaluate safety with regards to type, frequency, and severity of adverse events (AEs) and adverse events of special interest (AESI) and their relationship to study treatment,
- To evaluate patient related outcomes, measured in terms of health-related quality of life (QoL) by EORTC QLQ-C30 and QLQ-CLL16 questionnaires.
Secondary objectives (Arm B group)
- Time to and number of patients reinitiating treatment,
- Time to treatment failure after reinitiated treatment.
Eligibility
- Inclusion Criteria:
- Documented relapsed/refractory CLL or SLL requiring treatment according to IWCLL criteria (no limitis on previous treatment lines; CD20 and steroids are not considered prior therapy lines).
- Age at least 18 years.
- Adequate bone marrow function defined as:
- Absolute neutrophil count (ANC) >0.75 x 10^9/L
- Platelet count >30,000 /μL 30 x 10^9/L.
- Hemoglobin >8.0 g/dL (5 mmol/L). Unless directly attributable to CLL infiltration of the bone marrow, proven by bone marrow biopsy
- Creatinine clearance (CrCL) ≥ 30ml/min calculated according to the modified formula of Cockcroft and Gault or directly measured with 24hr urine collection.
- Adequate liver function as indicated
- Serum aspartate transaminase (AST) or alanine transaminase (ALT) ≤ 3.0 x upper limit of normal (ULN)
- Bilirubin ≤1.5 x ULN (unless bilirubin rise is due to Gilbert's syndrome or of nonhepatic origin)
- Prothrombin time (PT)/International normal ratio (INR) <1.5 x ULN and PTT (activated partial thromboplastin time [aPTT]) <1.5 x ULN (unless abnormalities are related to coagulopathy or bleeding disorder).
- Negative serological testing for hepatitis B (HBsAg negative and anti-HBc negative; patients positive for anti-HBc may be included if PCR for HBV DNA is negative and HBV-DNA PCR is performed every month until 12 months after last dose), negative testing for hepatitis C RNA within 42 days prior to registration.
- WHO/ECOG performance status 0-3 (appendix C), stage 3 only if attributable to CLL.
- Negative pregnancy test at study entry (for women of childbearing potential).
- Male and female subjects of reproductive potential must agree to use both a highly effective method of birth control (e.g. implants, injectables, combined oral contraceptives, some intrauterine devices [IUDs], complete abstinence , or sterilized partner) and a barrier method (e.g., condoms, cervical ring, sponge, etc.) during the period of therapy and for 90 days after the last dose of study drug.
- Ability and willingness to provide written informed consent and to adhere to the study visit schedule and other protocol requirements.
- Written informed consent.
Eligibility for randomization
- MRD negative peripheral blood and bone marrow at day 15 of cycle 15.
- At least PR after cycle 15.
Eligibility for Ibrutinib monotherapy in MRD-positive patients
- MRD positive peripheral blood and/or bone marrow at day 15 of cycle 15.
- At least SD after cycle 15
- Exclusion Criteria:
- Any prior therapy with ibrutinib and/or venetoclax.
- Transformation of CLL (Richter’s transformation).
- Patients with a history of confirmed progressive multifocal leukoencephalopathy (PML).
- Malignancies other than CLL currently requiring systemic therapies or not being treated in curative intention before or showing signs of progression after curative treatment.
- Known allergy to xanthine oxidase inhibitors and/or rasburicase if no other appropriate prevention of tumorlysis is considered feasible by the treating physician.
- Known bleeding disorders (e.g., von Willebrand’s disease or hemophilia).
- Uncontrolled or active infection.
- Patients requiring treatment with a strong cytochrome P450 (CYP) 3A inhibitor (seeappendix K). or anticoagulant therapy with warfarin or phenoprocoumon or other vitamin K antagonists.
Please note: Patients being treated with NOACs can be included, but must be properly informed about the potential risk of bleeding under treatment with ibrutinib.
- History of stroke or intracranial hemorrhage within 6 months prior to registration.
- Major surgery within 28 days prior to registration.
- Use of investigational agents which might interfere with the study drug within 28 days prior to registration.
- Vaccination with live vaccines within 28 days prior to registration
- Steroid therapy within 7 days prior to registration, with the exception of inhaled steroids for asthma, topical steroids, steroids up to 25 mg of prednisolone daily to control autoimmune phenomenon’s, or replacement/stress corticosteroids.
- Pregnant women and nursing mothers.
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; Use goto eCRF button > select the [Patient tab] and click the [Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31 (0)10 704 1028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31 (0)10 704 1560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Local patient code (optional)
- Sex
- Year of birth
- Date written informed consent
- Specific items patient gives consent for (see ICF)
- Eligibility criteria
Interim analysis V (Q2-2020)
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.