HOVON HO158 CLL
Main info
- Identifier:
- HO158 CLL
- Sponsor:
- HOVON
- Age:
- >= 18
- Stage:
- 1st Line
- Echelon:
- Level D
- Included patients:
-
85(of 75)
- Active sites:
-
18(of 26)
- Title:
First line treatment with VeNEtoclaX and ibruTinib induction followed by obinutuzumab intenSificaTion Exclusively in CLL/SLL Patients not in complete remission and/or with detectable bone marrow minimal residual disease (NEXT STEP trial)
Timeline
Flow
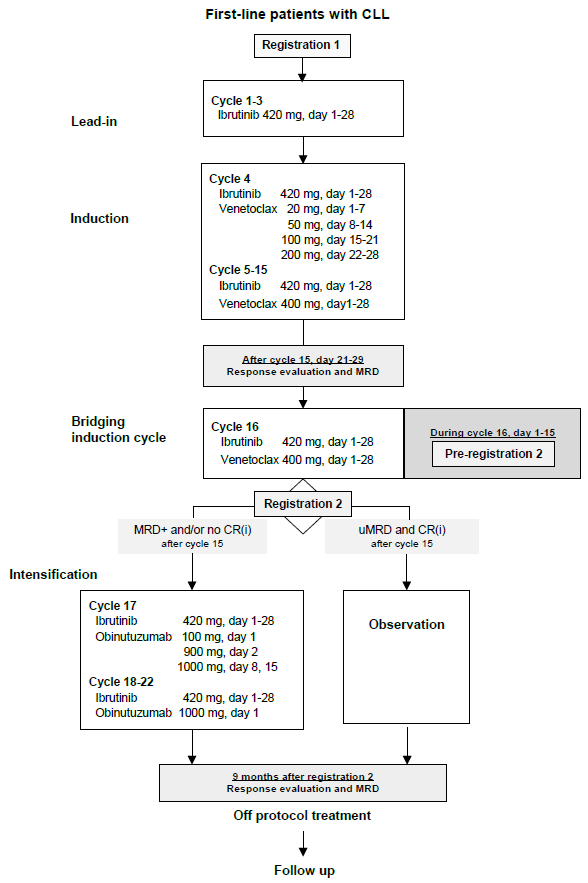
Details
- Phase:
- Select:
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
Primary objective
- To evaluate the efficacy of 6 cycles ibrutinib/obinutuzumab in converting patients who are not in CR or who have detectable MRD on combination ibrutinib and venetoclax in uMRD (BM) CR
Secondary objectives
- To explore the kinetics of CLL clearance on protocol and in follow up within the different compartments (PB, BM, LN) by regular test methods (MRD, PET-CT and BM)
- To measure the conventional response and outcomes of the combination ibrutinib/venetoclax and intensification with ibrutinib/obinutuzumab
- To evaluate prognostic parameters for response on ibrutinib/venetoclax and intensification with ibrutinib/obinutuzumab
- To evaluate safety of ibrutinib/venetoclax and intensification with ibrutinib/obinutuzumab
Exploratory objective
- To explore kinetics of CLL clearance on protocol and in follow up within different compartments (blood, BM, LN) by novel test methods (MRD, PET-CT, fine needle aspiration (FNA) of LN)
- To evaluate the impact on immunity (e.g. expression of CD20 and CXCR4/CD5) of ibrutinib/venetoclax and intensification with ibrutinib/obinutuzumab
- To evaluate quality of life (QoL) during and after ibrutinib/venetoclax and intensification with ibrutinib/obinutuzumab
Eligibility
- Inclusion Criteria:
- Documented CLL or SLL requiring treatment according to IWCLL criteria33, including minimal required markers (CD5/CD19/CD23 triple positive with light chain restriction);
- WHO performance status 0-3 (appendix C), stage 3 only if attributable to CLL/SLL;
- No prior treatment for CLL/SLL;
- Previous treatment with anti-CD20 antibody for another indication is allowed;
- Age at least 18 years;
- Adequate BM function defined as:
- Hb > 5 mmol/l or Hb > 8 g/dL
- Absolute neutrophil count (ANC) ≥ 0.75 x 10^9/L or 750/μL
- Platelet count ≥ 50 x 10^9/L or 50,000 /μL
Unless directly attributable to CLL/SLL infiltration of the BM, proven by BM biopsy;
- Estimated Glomerular Filtration Rate (eGFR) (MDRD) or estimated creatinine clearance (CrCl) ≥ 30ml/min (Cockcroft-Gault appendix E);
Please note: in case eGFR or CrCl is <50ml/min the patient needs to be considered high risk for TLS
- Adequate liver function as indicated:
- Serum aspartate transaminase (ASAT) and alanine transaminase (ALAT) ≤ 3.0 x upper limit of normal (ULN)
- Bilirubin ≤1.5 x ULN (unless bilirubin rise is due to Gilbert's syndrome or of nonhepatic origin);
- Prothrombin time (PT)/International normal ratio (INR) <1.5 x ULN and activated partial thromboplastin time (aPTT) <1.5 x ULN;
- Negative serological testing for hepatitis B virus (Hepatitis B surface antigen (HBsAg) negative and hepatitis B core antibody (anti-HBc) negative) and hepatitis C virus (hepatitis C antibody). Subjects who are positive for hepatitis B core antibody, hepatitis B surface antigen, or hepatitis C antibody must have a negative PCR result before enrollment. Those who are PCR positive will be excluded;
- Ability and willingness to adhere to the study visit schedule and other protocol requirements;
- Patient is capable of giving informed consent;
- Written informed consent.
- Exclusion Criteria:
- Transformation of CLL (Richter’s transformation);
- Malignancies other than CLL/SLL currently requiring systemic therapy or not being treated in curative intention or showing signs of progression after curative treatment;
- Patient with CNS involvement
- Known allergy to xanthine oxidase inhibitors and/or rasburicase;
- Intolerance of exogenous protein administration;
- History of severe allergic or anaphylactic reactions to humanized or murine monoclonal antibodies. Known sensitivity or allergy to murine products;
- Active fungal, bacterial, and/or viral infection that requires systemic therapy;
Please note: active controlled as well as chronic/recurrent infections are at risk of reactivation/infection during treatment (see section 9.2.3.1);
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled: infection, auto-immune hemolysis, immune thrombocytopenia, diabetes, hypertension, hyperthyroidism or hypothyroidism etc.);
- Patients known to be HIV-positive;
- Patient requiring treatment with a strong cytochrome P450 (CYP) 3A inhibitor (see appendix K) or anticoagulant therapy with warfarin or phenoprocoumon or other vitamin K antagonists;
Please note: Patients being treated with DOACs apixaban, edoxaban or rivaroxaban can be included, but must be properly informed about the potential risk of bleeding under treatment with ibrutinib. Treatment with dabigatran should be avoided, due to risk of toxicity based on P-gp substrate (see appendix K)
- History of stroke or intracranial hemorrhage within 6 months prior to registration;
- Severe cardiovascular disease (arrhythmias requiring chronic treatment, congestive heart failure or symptomatic ischemic heart disease) (CTCAE grade III-IV, see appendix D);
- Severe pulmonary dysfunction (CTCAE grade III-IV, see appendix D);
- Patient with Child Pugh C
- Severe neurological or psychiatric disease (CTCAE grade III-IV, see appendix D);
- Vaccination with live vaccines within 28 days prior to registration;
- Use of any other experimental drug or therapy within 28 days prior to registration
- Major surgery within 28 days prior to registration;
- Steroid therapy within 10 days prior to registration, with the exception of inhaled steroids for asthma, topical steroids, steroids up to 20 mg of dose equivalents of prednisolone daily to control autoimmune phenomenon’s, or replacement/stress corticosteroids;
- Pregnant women and nursing mothers;
- Fertile men or women of childbearing potential unless: (1) surgically sterile or ≥ 2 years after the onset of menopause, and/or (2) willing to use a highly effective contraceptive method such as oral contraceptives, intrauterine device, sexual abstinence or barrier method of contraception in conjunction with spermicidal jelly during study treatment and in female patients for 3 months after end of induction treatment and 18 months after end of treatment with obinutuzumab and male patients for 6 months after end of treatment;
- Current participation in other clinical trial;
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Registration 1: Eligible patients should be registered before start of treatment. Patients need to be registered at HOVON Data Center by one of the following options:
- HOVON Data Center registration database www.hovon.nl). Account for registration and a registration manual can be requested at HOVON Data Center.
- By sending the completed registration CRF by e-mail to hdc@erasmusmc.nl, Monday through Friday from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Sex
- Age
- Date written informed consent
- Specific items patient gives consent for (see ICF for biobank)
- Eligibility criteria
- Binet classification
- CIRS score
All eligibility criteria will be checked.
Each patient will be given a unique patient study number (a sequence number by order of enrolment in the trial). Patient study number will be given immediately the online registration database or phone and confirmed by email.
(Pre-) registration 2
Pre-registration 2 should be performed during cycle 16, latest at day 15, for confirmation of IWCLL response and collection of MRD results by (co)PIs. Confirmation of response is required for continuation of treatment or observation.
Each center will receive a reminder email for pre-registration before start cycle 16.
Patients need to be pre-registered at HOVON Data Center by the following option:
- HOVON Data Center registration database (www.hovon.nl).
The following information will be requested at pre-registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- IWCLL response conclusion end of cycle 15
- LN > 1.5 cm in longest diameter on CT yes/no
- Spleen > 13 cm yes/no
- Abnormal liver yes/no
- Constitutional symptoms yes/no
- Peripheral blood lymphocytes end of cycle 15
- Hemoglobin end of cycle 15
- Thrombocytes end of cycle 15
- Normocellular BM, no CLL cells, no B-lymphoid nodules yes/no
- Anonymized original reports of blood values, CT scan and BM performed at end of cycle 15 (upload in ALEA)
- Date start of cycle 16.
Within 1 week after pre-registration 2 but no later than day 21 of cycle 16, confirmation of IWCLL response and MRD result will be sent by email to the center.
Pre-registration 2 will result in start of intensification treatment or observation. Day 1 of intensification or observation (29 days after start cycle 16) will be defined as registration 2, and will be the reference date for following evaluations.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.