HOVON HO171 AML
Main info
- Identifier:
- HOVON 171 AML
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- >= 18
- Stage:
- 1st Line
- Echelon:
- Level C-HIC&C-SCT
- Included patients:
-
124(of 142)
- Active sites:
-
19(of 19)
- Title:
A single arm phase II trial to assess cobicistat boosted venetoclax in combination with azacitidine (sc) in patients with newly diagnosed acute myeloid leukaemia (AML) who are not considered candidates for intensive treatment regimens
Timeline
News
30JUN2026: Laboratory Manual v8, 24JUN2026 is available. Be also referred to the DHL booking form and fillable form to order DHL shipping boxes.
20APR2026: Biobank ICF_NL_v1.2 18FEB2026 and Biobank protocol are available.
19FEB2026: Protocol v5.1_02DEC2025; ICF Main-NL_v5.2_15DEC2025 and ICF pre-study_NL_v3.1 02DEC2025 are available.
13DEC2024:
As of 13DEC2024, the run-in phase of the HO171 has been completed. From this point onwards, all patients that are included in the study will participate in the extension phase.
03JUL2024:
Substantial Amendment 1 has been approved and been shared with the participating sites for implementation.
17JAN2024:
The first site (NL-Groningen-UMCG) has been activated today for recruitment.
25OCT2023
Study is approved in CTIS.
Flow
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- HOVON Monitoring Visit
- Objectives:
Primary objectives:
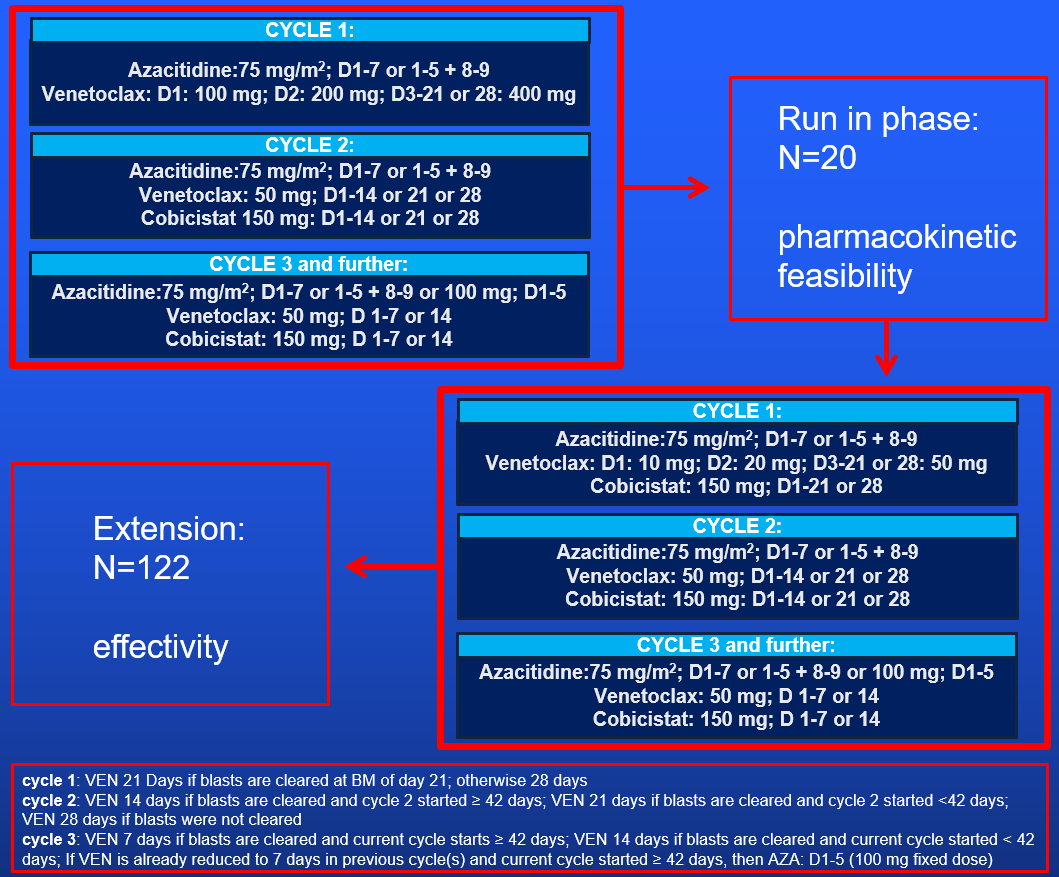
Run-in phase- To evaluate the pharmacokinetic (PK) equivalence of cobicistat boosted venetoclax and unboosted venetoclax (PK cycle 1 vs PK cycle 2).
Extension phase
- To assess the practical applicability and the therapeutic effect of a defined schedule of cobicistat added to venetoclax and azacitidine (AZA/VEN/COBI) on overall survival (OS) in adult patients with newly diagnosed AML considered ineligible for intensive chemotherapy.
Secondary objectives:
Run in- To assess venetoclax and cobicistat CL, Cmax, Tmax, Cmin, AUC 0-24.
Extension phase
- To determine the efficacy profile: response rate (CR, CRi, CR/CRi, CRh, CR/CRh, CRMRD-, CR/CRiMRD-, CR/CRhMRD-, and MLFS), event-free survival (EFS) and relapse-free survival (RFS) associated with AZA/VEN/COBI.
- To evaluate safety and toxicity of AZA/VEN/COBI.
- To assess the impact of cytogenetic and molecular genetic factors on response and survival endpoints.
- To compare the OS of AZA/VEN/COBI treated patients with a real-world data cohort treated during the same period and monitored by the Dutch Cancer registry.
- To assess the exposure-response and exposure-toxicity relation of venetoclax in patients with AML.
- To assess adherence to venetoclax and cobicistat.
- To assess cost-savings on venetoclax drug costs (corrected for cobicistat costs) compared to the standard dosing schedule.
Eligibility
- Inclusion Criteria:
- Patients with: a diagnosis of AML and related precursor neoplasms according to ICC-2022 classification (excluding acute promyelocytic leukaemia) (Appendix A). Patients may have had previous treatment with erythropoiesis stimulating agents (ESA) for an antecedent phase of MDS. ESAs must be stopped at least two weeks before registration.
- Patients 18 years and older who are considered not fit for intensive chemotherapy (judged by treating physician) or who decline the option of intensive chemotherapy.
- WHO performance status 0, 1 or 2 (Appendix E).
- Adequate renal and hepatic functions unless clearly disease related as indicated by the following laboratory values:
- Adequate renal function as demonstrated by a creatinine clearance ≥ 30 mL/min; calculated by the Cockcroft Gault formula or measured by 24 hours urine collection.
- Serum bilirubin ≤ 3 x upper limit of normal (ULN), unless considered AML-related or due to Gilbert’s syndrome.
- Alanine transaminase (ALT) ≤ 3 x ULN, unless considered AML-related.
- Male subjects who are sexually active, must agree, from Study Day 1 until at least 90 days after the last dose of study drug, to practice the protocol specified contraception. Male subjects must agree to refrain from sperm donation from initial study drug administration through at least 90 days after the last dose of study drug.
- Female subjects must be either postmenopausal defined as: Age >55 years with no menses for ≥12 months, without an alternative medical cause. OR willing and able to use adequate contraception during and until 180 days after the last protocol treatment.
- Written informed consent.
- Patient is capable of giving informed consent.
- Patient agrees not to participate in another interventional study while on protocol treatment without approval of the (co-) Principal Investigator.
- Exclusion Criteria:
- Acute promyelocytic leukemia.
- Myelodysplastic syndrome (MDS).
- Patients previously treated for AML or MDS (any anti-leukemic therapy including investigational agents; excluding: 1) erythropoiesis stimulating agents (ESAs); 2) hydroxyurea (hydroxyurea is allowed for the control of peripheral leukemic blasts in patients with leukocytosis).
- Diagnosis of any previous or concomitant malignancy is an exclusion criterion:
- except when the patient successfully completed treatment (chemotherapy and/or surgery and/or radiotherapy) with curative intent for this malignancy at least 24 months prior to registration;
- except for basal and squamous cell carcinoma of the skin or in situ carcinoma of the cervix.
- Blast crisis of chronic myeloid leukemia.
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, pulmonary disease etc.).
- Cardiac dysfunction as defined by:
- Myocardial infarction within the last 3 months of study entry, or
- Reduced left ventricular function with an ejection fraction < 40% as measured by MUGA scan or echocardiogram, or
- Unstable angina or New York Heart Association (NYHA) grade IV congestive heart failure (see Appendix G), or
- Unstable cardiac arrhythmias.
- History of stroke or intracranial haemorrhage within 6 months prior to registration.
- Symptomatic central nervous system (CNS) leukemia (NO routinely lumbar puncture required to investigate CNS involvement).
- History of non-compliance to medical regimens or considered unreliable with respect to compliance.
- Senile dementia, mental impairment or any other psychiatric disorder that prohibits the patient from understanding and giving informed consent.
- Current concomitant chemotherapy, radiation therapy, or immunotherapy; other than hydroxyurea.
- Any psychological, familial, sociological or geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
- Unreplaceable use of strong inhibitors or inducers of CYP3A or CYP3A/p-GP substrates with a narrow therapeutic window (e.g., cobicistat or ritonavir for HIV treatment). Please check with Appendix I.
- Intolerability, contra-indication or allergy to one of the study drugs.
Exclusion for the run-in phase only:
- Patient with a strong indication for IFD prophylaxis (or treatment)
Registration Details
Patients who are possibly eligible for this trial should first be registrated in the screeningsdatabase. This can only be done after the patient has provided a written informed consent for the pre-study. This pre-study registration is done through the screening database.
Eligible patients should be registered before start of treatment. Patients need to be registered at HOVON by one of the following options:
- HOVON registration database (see HOVON website www.hovon.nl for the most recent link to ALEA). Account for registration and a registration manual can be requested at HOVON.
- By sending the completed registration/randomization CRF by e-mail to hovon@erasmusmc.nl. Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Sex
- Year of birth
- Date written informed consent
- Specific items patient gives consent for (see ICF)
- Eligibility criteria
All eligibility criteria will be checked automatically.
Each patient will be given a unique patient study number (a sequence number by order of enrolment in the trial).
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.