HOVON HO89 AML
Main info
- Identifier:
- HOVON 89 MDS
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- >= 18
- Stage:
- 1st Line
- Echelon:
- Level D
- Included patients:
-
200(of 200)
- Active sites:
-
60(of 46)
- Title:
A Phase II randomized multicenter study to assess the efficacy of lenalidomide with or without erythropoietin and granulocyte-colony stimulating factor in patients with low and intermediate-1 risk myelodysplastic syndrome.
Timeline
Flow
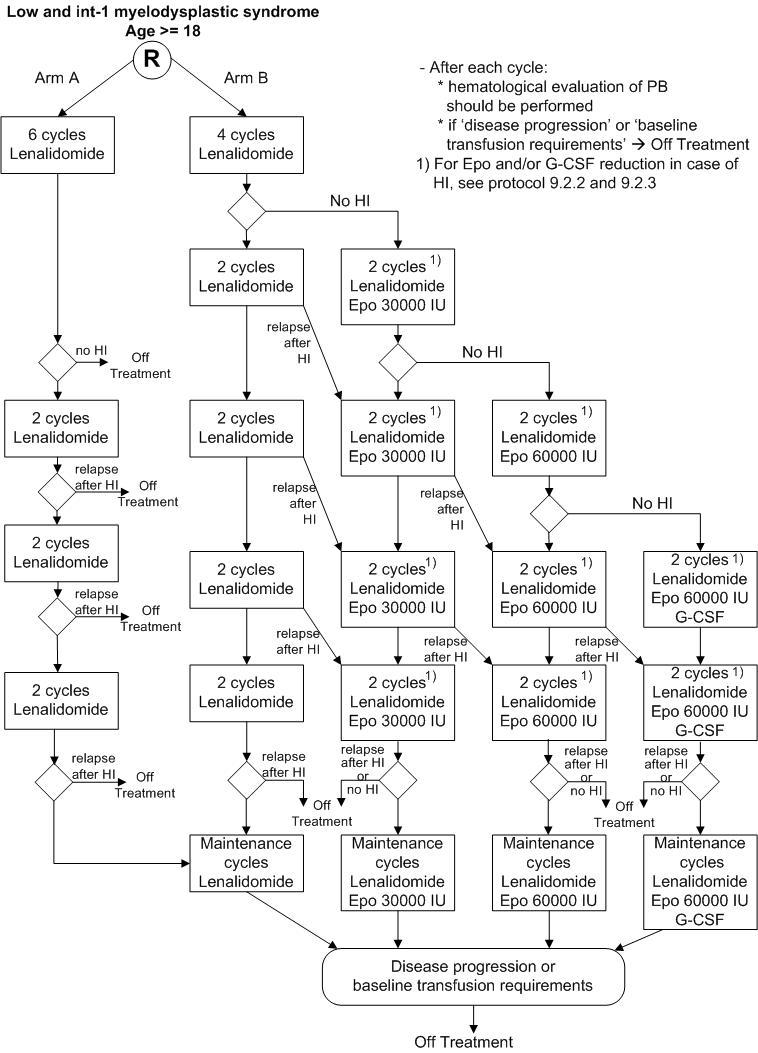
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- Not any more
- Objectives:
Primary objective
- To evaluate the efficacy of lenalidomide (RevlimidTM) in low/int-1 risk MDS with or without a treatment with Epo (NeoRecormonTM)/G-CSF (NeupogenTM) in terms of hematological improvement (HI) as defined by the modified response criteria of the IWG for MDS [40], see appendix C.
Secondary objectives
- To evaluate the safety and tolerability of lenalidomide (RevlimidTM) in low/int-1 risk MDS with or without Epo (NeoRecormonTM)/G-CSF (NeupogenTM)
- Time-to-HI and duration-of-HI
- The number of given treatment cycles per patient and for arm B the number of patients receiving Epo and/or G-CSF
- The response rate (in terms of CR, PR, including cytogenetic response according to the modified response criteria of the IWG for MDS [40], see appendix C)
- Progression-Free-Survival (i.e. time from registration to disease progression, including progression to leukemia, or death from any cause)
- Transfusion requirements of red blood cells
Eligibility
- Inclusion Criteria:
- Patients with MDS classified as
- RA, RARS and RAEB (with <10% myeloid blasts), CMML (with <10% myeloid blasts), according to FAB (see appendix A3) or
- RA, RARS, RCMD, RCMD-RS, RAEB-1, MDS-U according to WHO (see appendix A1) or
- patients with MPD/MDS (CMML-1 according to WHO) with a WBC ≤ 12x10^9/l (see appendix A2)
with an IPSS ≤ 1.0 (see appendix B1)
- Hb ≤ 6.2 mmol/l (10.0 g/dl)
or Hb ≤ 7.2 mmol/l and ANC ≤ 1.0x10^9/l
or red blood cell transfusion dependent (≥ 2 units RBC during at least 8 weeks; units must be given for a Hb ≤ 5.6 mmol/l)- Age ≥ 18 years
- WHO performance status 0-2 (see appendix D)
- Patient not previously treated with Epo/G-CSF, or failure of response or relapse after hematological improvement or disease progression to maximal RAEB-1 after previous therapy with Epo/G-CSF
- Serum creatinin < 150 μmol/l
- Serum billirubin < 25 μmol/l and ASAT, ALAT and Alkaline phosphatase < 2.5 times the upper limit of normal, except if related to disease
- The patient must give written informed consent
- Negative pregnancy test within 7 days prior to start of study drug, if applicable.
- Patient (all men, pre-menopausal women) agrees to use adequate contraceptive methods.
- Serum erythropoietin level
- > 200 U/l or
- ≤ 200 U/l if failure of response or loss of hematological improvement or disease progression to maximal RAEB-1 after prior standard therapy with Epo/G-CSF; Epo/G-CSF should be stopped at least 1 month before randomization.
- Patients with MDS classified as
- Exclusion Criteria:
- Severe cardiac, pulmonary, neurologic, metabolic or psychiatric diseases or active malignancies.
- Anemia due to other causes than MDS including iron, B12 and folate deficiencies, autoimmune hemolysis and/or paroxysmal noctural hemoglobinuria (PNH)
- Hypoplastic MDS
- High predictive score (score 0 or 1) to respond on standard treatment with Epo/G-CSF according to guidelines; see appendix H
- Active uncontrolled infection
- Absolute neutrophil count (ANC) < 0.5x10^9/l
- Patients dependent on platelet transfusions or with platelet counts < 25x10^9/l or patients with active bleeding
- Patients treated with biological response modifiers (i.e. growth factors, immunosuppressive agents and/or chemotherapy) within 1 month prior to randomization
- Lactating women
- Prior treatment with lenalidomide
- Prior CTCAE ≥ grade 3 allergic reaction/hypersensitivity to thalidomide
- Prior CTCAE ≥ grade 3 rash/blistering while taking thalidomide
- Prior CTCAE ≥ grade 3 allergic/hypersensitivity to Epo and/or G-CSF
Registration Details
Eligible patients should be randomized before start of treatment. Patients need to be registered at the HOVON Data Center of the Erasmus MC Rotterdam – location Daniel den Hoed, via the Internet via TOP (Trial Online Process; https://www.hdc.hovon.nl/top) or by phone call: +31.10.7041560 or fax +31.10.7041028, Monday through Friday, from 09:00 to 17:00 CET. A logon to TOP can be
requested at the HOVON Data Center for participants.
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Sex
- Date of birth
- Date written informed consent
- ‘Risk Management Program’ is discussed with patient
- Approval for central tissue review
- Approval for blood and/or bone marrow storage for scientific research
- Presence of cytogenetic abnormalities
- MDS diagnosis (FAB and/or WHO)
- Eligibility criteria
Final analysis for manuscript (Q2/Q3-2020)
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.