HOVON HO92 AML
Main info
- Identifier:
- HOVON 92 AML
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- 18-65
- Stage:
- 1st Line
- Echelon:
- Limited Site Selection
- Included patients:
-
148(of 800)
- Active sites:
-
28(of 34)
- Title:
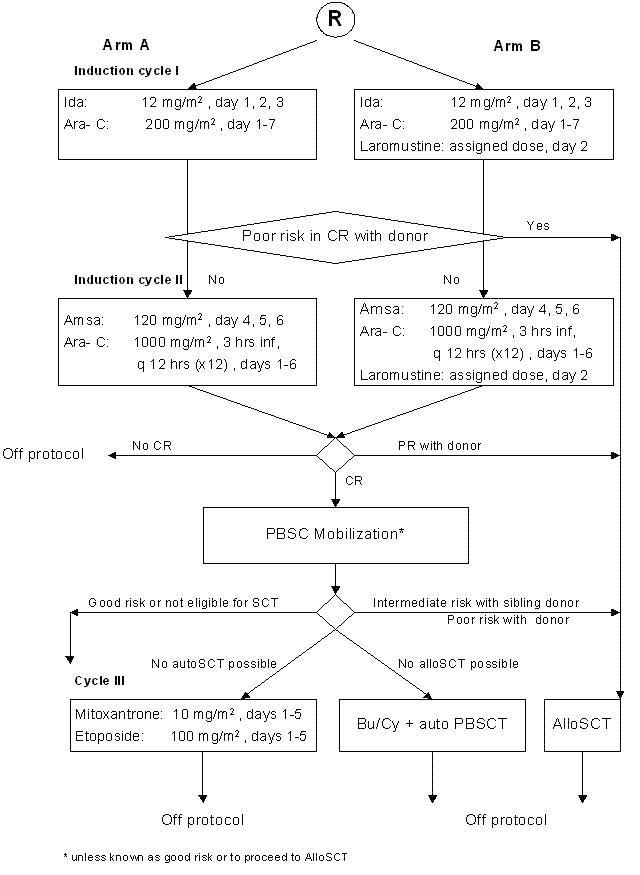
Randomized study to assess the added value of Laromustine in combination with standard remission-induction chemotherapy in patients aged 18-65 years with previously untreated acute myeloid leukemia (AML) or myelodysplasia (MDS) (RAEB with IPSS ≥ 1.5).
Timeline
News
HOVON 92 AML / SAKK 30/08: please note, the study has been closed.
Updated documents (in reversed chronological order of date replacement):
- New CRF version of 10 March 2009. Changes: addition on DLT form and PDF correct conversion of symbols.
- PI & ICF amendment(s) new version that clearifies the fact that there were two versions of Part A and Part B submitted to the cMETC.
- Amendment 1 replacements/additions:
- Protocol (now version 02dec08)
- Protocol amendment(s)
- CRFs (now version 30jan09)
- CRF instructions (now version 24feb09)
- PI & ICF amendment(s)
- MEC approval (d.d. 27feb09)
- Signature page of protocol
- Registration (&randomization) form (now version 30jan09)
- HO92PIF_part A_AM1_30jan09
- HO92PIF_partB_AM1_30jan09
- HO92PIF_PK_AM1_02dec08
- DLT form_30jan09
- Instructions for laboratory investigations HO92_Instructions for laboratory investigations_v04MAR09 has been updated to give clearer information.
- Statement of expenses form for NLsites HO92_declarationform_NLsites_v03MAR09 has been updated.
- Document named HO92_Questions_and_Answers_v28jan09 has been updated.
- Document named ho92_vpk_werkdocument_general_v06nov08 has been updated.
- Document named HO92_Questions_and_Answers_v23jan09 has been updated.
- CKTO approval has been received, the document can be found below 'forms for local submission'
- ho92_pk_vpk_werkdocument_general_v07nov08 instead of version dd28OCT08 which had incorrect data on sheet regarding Arm B bloodsamples.
- MEC Correspondence has been replaced by a new version. New version also includes email send by cMETC on 28AUG08 and response by PI dd 03SEP08.
Flow
Details
- Phase:
- Prospective randomized Phase II/III study
- Monitoring Type:
- Not any more
- Objectives:
Primary objectives
For part A of the study:- To determine the feasibility of Laromustine when given at three possible dose levels together with standard induction cycles I and II in patients with AML/ RAEB with IPSS ≥1.5 in a prospective comparison to standard induction cycles I and II without Laromustine
For part B of the study:
- To evaluate the effect of Laromustine at the selected feasible dose level when combined with remission induction chemotherapy cycles I and II as regards clinical outcome (“event-free survival”) in comparison to remission induction cycles I and II with no addition of Laromustine in a phase III study
Secondary objectives
For part A of the study:- To evaluate the pharmacokinetics of Laromustine in the combination with cytarabineidarubicin remission induction chemotherapy in a selection of patients at different dose levels of Laromustine as well as in a limited number of controls
- To investigate the clinical efficacy of Laromustine in combination with remission induction chemotherapy cycles I and II with regard to complete remission rate at different dose levels of Laromustine
For part B of the study:
- To investigate the clinical efficacy of Laromustine with regard to the complete remission rate, disease free survival (DFS), risk of relapse and overall survival (OS) when combined with remission induction chemotherapy cycles I and II in all patients
- To investigate the clinical efficacy of Laromustine when combined with remission induction chemotherapy cycles I and II in molecularly and cytogenetically distinguishable subsets with regard to the complete remission rate, disease free survival (DFS), risk of relapse and overall survival (OS)
- To investigate the tolerance and toxicity of Laromustine in combination with remission induction chemotherapy cycles I and II
- To evaluate the pharmacokinetics of Laromustine and cytarabine-idarubicine remission induction chemotherapy in a limited number of patients in both treatment arms
- To assess the effect of Laromustine on peripheral CD34 cell numbers for autologous peripheral blood transplantation
- To determine the prognostic value of molecular markers and gene expression profiles of the leukemia assessed at diagnosis
- To evaluate the treatment effects according minimal residual disease (MRD) measurements following therapy by standardized sampling of marrow/blood
- To evaluate the outcome of allogeneic sibling or unrelated donor SCT and autologous SCT in cytogenetically and molecularly defined and prognostic subgroups of patients.
Eligibility
- Inclusion Criteria:
- Age 18-65 years, inclusive
- Subjects with
- a cytopathologically confirmed diagnosis of AML according WHO classification (excluding acute promyelocytic leukaemia) or
- a diagnosis of refractory anemia with excess of blasts (RAEB) and IPSS score ≥1.5 or
- patients with therapy-related AML/RAEB or
- patients with biphenotypic leukemia (Appendices A1 and A2).
- WHO performance status 0, 1 or 2 (see Appendix I)
- Written informed consent
- Exclusion Criteria:
- During part A of the study patients with a good risk AML, if already known at randomisation. These patients will be treated outside the study according to the control arm.
- Acute promyelocytic leukaemia
- Previous treatment for AML or RAEB, except hydroxyurea
- Impaired hepatic or renal function as defined by:
- ALT and/or AST > 3 x Upper Limit of Normal (ULN), or
- Bilirubin > 3 x ULN, or
- Serum creatinine> 3 x ULN (after adequate hydration), unless these are most likely caused by AML organ infiltration,
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, pulmonary disease etcetera),
- Cardiac dysfunction as defined by:
- Myocardial infarction within the last 6 months of study entry, or
- Reduced left ventricular function with an ejection fraction < 50% as measured by MUGA scan or echocardiogram (another method for measuring cardiac function is acceptable), or
- Unstable angina, or
- Unstable cardiac arrhythmias
- Pregnant or lactating females
- Impossibility to stop Disulfiram (Antabuse) and metronidazol (Flagyl) 24 hours prior to study treatment. (Please note that this medication must be stopped 24 hours prior to study treatment.)
- Unwilling or not capable to use effective means of birth control
Registration Details
Eligible patients should be randomized before start of induction treatment. Patients need to be randomized at the HOVON Data Center of the Erasmus MC Rotterdam – location Daniel via the Internet via TOP (Trial Online Process; https://www.hdc.hovon.nl/top) or by phone call: +31.10.7041560 or fax +31.10.7041028 Monday through Friday, from 09:00 to 17:00 CET. A logon to TOP can be requested at the HOVON Data Center for participants.
The following information will be requested at randomization:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Patient’s initials or code
- Sex
- Date of birth
- Date of diagnosis of AML or RAEB
- Date written informed consent
- Eligibility criteria
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.