GSK HO98 NHL
Main info
- Identifier:
- HO98 NHL
- Sponsor:
- GSK
- Working group party:
- Lymphoma
- Age:
- >= 18
- Stage:
- 2nd Line
- Included patients:
-
42(of 447)
- Active sites:
-
13(of 170)
- Title:
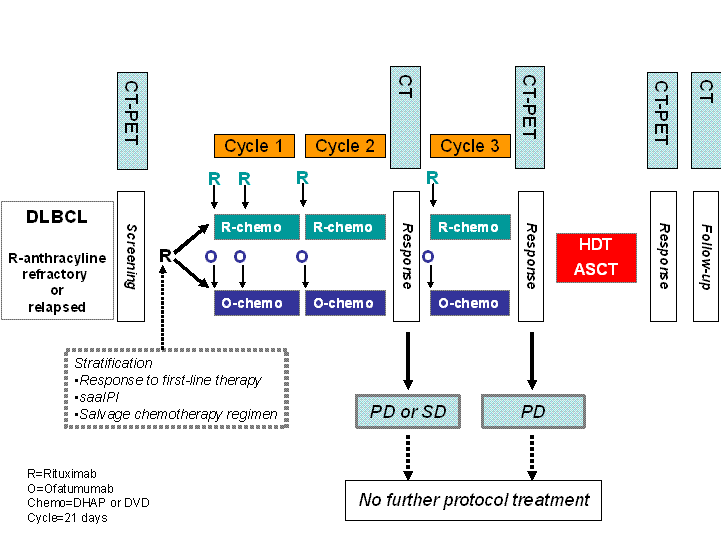
Ofatumumab versus Rituximab Salvage Chemoimmunotherapy followed by ASCT in Relapsed or Refractory DLBCL
Timeline
News
Site documents will be provided by GSK . Please contact Claudia Leemereise (claudia.n.leemereise@gsk.com)
November 16, 2013: Study is closed for entry of new patients
July 3, 2012: updated protocol available
Flow
Details
- Phase:
- Prospective randomized Phase III study
- Monitoring Type:
- Objectives:
Primary objective:
To evaluate the progression-free survival (PFS) in subjects receiving ofatumumab in addition to salvage chemotherapy (O-chemo) compared to subjects receiving rituximab in addition to salvage chemotherapy (R-chemo).Secondary objectives: To evaluate the following in subjects receiving O-chemo compared to subjects receiving R-chemo:
- Overall and complete response rate after salvage chemoimmunotherapy.
- Overall and complete response rate three months after ASCT.
- Event-free survival.
- Overall survival.
- Number of subjects with inadequate mobilisation of autologous stem cells (<2.0 million CD34+ cells/kg) prior to administration of high dose therapy (HDT)
- Number of subjects completing ASCT.
- Changes in patient health-related quality of life (HRQL) measures
- Incidence, severity of adverse events, serious adverse events and other safety parameters.
- Time to neutrophil and platelet recovery after each cycle of salvage chemotherapy and time to engraftment after HDT/ASCT.
Exploratory objectives: To evaluate the following in subjects receiving O-chemo compared to subjects receiving R-chemo:
- Pharmacogenetics profile associated with response to ofatumumab.
- Biomarker profiles of DLBCL/grade 3b FL tissue samples to identify factors that may influence biological and clinical responses to ofatumumab and/or associated with the development or progression of DLBCL/grade 3b FL or medically related conditions.
Eligibility
- Inclusion Criteria:
- CD20 positive DLBCL or grade 3b follicular lymphoma (FL) at original diagnosis. If an evaluable biopsy or fine needle aspiration (FNA) is performed prior to enrolment to the study it must confirm CD20 positive DLBCL or grade 3b FL. Note: If evidence
emerges that the binding of the immunohistochemical antibody to CD20 can be blocked by rituximab, demonstration of CD20 positivity in the repeat biopsy/FNA will not be required.
- Refractory to, or relapsed following, first-line treatment with rituximab concurrently with anthracycline- or anthracenedione-based chemotherapy.
Relapse is defined as:
- Biopsy (preferred) or FNA confirmed DLBCL or grade 3b FL after a complete response (CR) or unconfirmed complete response (CRu). However, for subjects relapsing during first-line treatment, biopsy/FNA reconfirmation of the lymphoma is recommended but not mandatory.
Subjects must have received rituximab concurrently with at least 6 cycles of chemotherapy. However, subjects with stage I/II disease will be eligible if they have received rituximab concurrently with at least 3 cycles of chemotherapy and definitive involved-field radiation therapy. Eligibility of subjects treated with an intensive chemotherapy regimen will be determined by HOVON.
Refractory disease must fulfill one of the following:
- continuing partial response (PR) from termination of first-line treatment. It is strongly recommended the lymphoma be reconfirmed by biopsy (preferred) or FNA, however, if these procedures are deemed to be inappropriate, then HOVON may determine eligibility following review of the imaging results and disease history.
Subjects must have received rituximab concurrently with at least 6 cycles of chemotherapy. However, subjects with stage I/II disease will be eligible if they have received rituximab concurrently with at least 3 cycles of chemotherapy and definitive involved-field radiation therapy. Eligibility of subjects treated with an intensive chemotherapy regimen will be determined by HOVON.
**continuing stable disease (SD) from termination of first-line treatment. Reconfirmation of the lymphoma by biopsy (preferred) or FNA is recommended but not mandatory.Subjects must have received rituximab concurrently with at least 3 cycles of chemotherapy. Eligibility of subjects treated with an intensive chemotherapy regimen will be determined by HOVON.
- progressive disease (PD). Biopsy or FNA reconfirmation of the lymphoma is recommended but not mandatory.
Note: Disease response to first-line treatment is recommended to be determined according to Revised Response Criteria for Malignant Lymphoma [Cheson, 2007] or International Workshop Response criteria for NHL [Cheson, 1999]. For guidance on
the adequacy of dosing of rituximab during first-line therapy refer to the SPM.- Baseline FDG-PET scans must demonstrate positive lesions compatible with CT defined anatomical tumor sites.
- CT scan showing at least:
- 2 or more clearly demarcated lesions/nodes with a long axis >1.5cm and short axis ≥1.0cm
OR
- 1 clearly demarcated lesion/node with a long axis >2.0cm and short axis ≥1.0cm.
- Age ≥18. For India only: age must also be ≤55 years.
- ECOG performance status 0, 1, or 2.
- Eligible for high dose chemotherapy and ASCT.
- Resolution of toxicities from first-line therapy to a grade that in the opinion of the investigator does not contraindicate study participation. The GSK Medical Monitor is available to discuss subject eligibility for all criteria.
- Signed written informed consent.
- Exclusion Criteria:
- Any previous cancer therapy for the lymphoma, with the exception of:
- First-line treatment with rituximab and an anthracycline- or anthracenedionebased chemotherapy.
- Monotherapy rituximab, dosed prior to first-line rituximab combined with chemotherapy, or as maintenance therapy.
- Radiotherapy as part of the first-line treatment plan.
- Radiotherapy to a limited field at a maximum dose of ≤10Gy to control lifethreatening symptoms.
- Prophylactic testicular radiotherapy for testicular lymphoma.
- Intrathecal chemotherapy for the prophylaxis of CNS disease.
- Received any of the following treatments within two weeks prior to start of study therapy (unless otherwise stated):
- Anti-cancer cytotoxics (e.g. alkylating agents, anti-metabolites, purine analogues)
- Radiotherapy unless it is to a limited field at a maximum dose of ≤10Gy to control life-threatening symptoms.
- Treatment with any known non-marketed drug substance or experimental therapy within 5 terminal half lives or 4 weeks prior to enrollment, whichever is longer, or currently participating in any other interventional clinical study unless in the opinion
of the investigator it does not contraindicate participation in this study.
- Planned post-randomisation glucocorticoid therapy, unless
- specified by the protocol
- administered in doses ≤1mg/kg/day prednisolone (or equivalent dose of other glucocorticoid-refer to the SPM for glucocorticoid equivalent doses)
- administered as inhalation therapy for mild COPD or asthma.
- History of significant cerebrovascular disease or event with significant symptoms or sequelae, unless in the opinion of the investigator it does not contraindicate participation in the study.
- Clinically significant cardiac disease including unstable angina, acute myocardial infarction within six months prior to randomisation, congestive heart failure (NYHA III-IV), a current LVEF of <40%, and arrhythmia unless controlled by therapy, with
the exception of extra systoles or minor conduction abnormalities, unless in the opinion of the investigator it does not contraindicate participation in the study.
- Significant concurrent, uncontrolled medical condition that in the opinion of the investigator contraindicates participation in this study.
- Known lymphoma involvement of the CNS.
- Known or suspected hypersensitivity to study treatments that in the opinion of the investigator contraindicates their participation.
- Known HIV positivity.
- Positive serology for Hepatitis B (HB) defined as a positive test for HBsAg. In addition, if negative for HBsAg but HBcAb positive (regardless of HBsAb status), a HB DNA test will be performed and if positive the subject will be excluded.
- Active hepatitis C infection.
- Chronic or current infectious disease requiring systemic antibiotics, antifungal, or antiviral treatment such as, but not limited to, chronic renal infection, chronic chest infection with bronchiectasis and tuberculosis.
- Other past or current malignancy within 2 years prior to randomization unless in the opinion of the investigator it does not contraindicate participation in the study. Subjects who have been free of malignancy for at least 2 years, or have a history of
completely resected non-melanoma skin cancer, or successfully treated in situ carcinoma, are eligible.
- Prior treatment with anti-CD20 monoclonal antibodies with the exception of rituximab.
- Screening laboratory values:
- platelets <100x10^9/L (unless due to lymphoma involvement of the bone marrow)
- neutrophils <1.5x10^9/L (unless due to lymphoma involvement of the bone marrow)
- creatinine >2.0 times upper normal limit (unless due to lymphoma or unless creatinine clearance >60mL/min)
- total bilirubin >1.5 times upper normal limit (unless due to lymphoma or a known history of Gilbert’s disease)
- ALT >2.5 times upper normal limit (unless due to lymphoma)
- alkaline phosphatase >2.5 times upper normal limit (unless due to lymphoma )
- Subjects known or suspected of being unable to comply with the study protocol.
- Pregnant or lactating women. Women of childbearing potential must have a negative pregnancy test at screening.
- Women of childbearing potential, including women whose last menstrual period was less than one year prior to screening, unable or unwilling to use adequate contraception from study start to one year after the last dose of protocol therapy.
Adequate contraception is defined as hormonal birth control, intrauterine device, double barrier method or total abstinence.
- Male subjects unable or unwilling to use adequate contraception methods from study start to one year after the last dose of protocol therapy.
The GSK Medical Monitor is available to discuss subject eligibility for all criteria.
- Any previous cancer therapy for the lymphoma, with the exception of:
Registration Details
Investigators or designated staff will telephone the GSK interactive voice response system (IVRS) called RAMOS (Registration And Medication Ordering System) to register and record subject activity. To randomize the subject, the study staff will enter the subject number and details of the subject’s stratification factors to obtain a randomization number and treatment group assignment. All calls to RAMOS are confirmed with a fax, which will be sent to the site on the completion of each call. Once a randomization number has been assigned it must not be re-assigned. Instructions for using the IVRS and determining proper stratification are provided in the SPM.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.