HOVON HO103 AML
Main info
- Identificatie:
- HOVON 103 AML
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- > 65
- Stadium:
- 1st lijn
- Echelon:
- Level C-HIC&C-SCT
- Included patients:
-
679(of 700)
- Active sites:
-
44(of 46)
- Title:
A program of randomized phase II multicenter studies to assess the tolerability and efficacy of the addition of new drugs to standard induction chemotherapy in AML and high risk myelodysplasia (MDS) (IPSS-R risk score > 4.5)in patients aged >= 66 years.
Timeline
News
Discontinuation of the study
An efficacy interim analysis for the HOVON 103 AML Selinexor study, has been performed according to protocol after the first 50 patients evaluable for induction treatment response in each treatment arm.
The results were reviewed by an independent DSMB and they advised to discontinue the study according to their opinion and in line with the guidelines in the protocol. The HOVON executive board and the principal investigator of the study support the conclusion to stop the study.
Flow
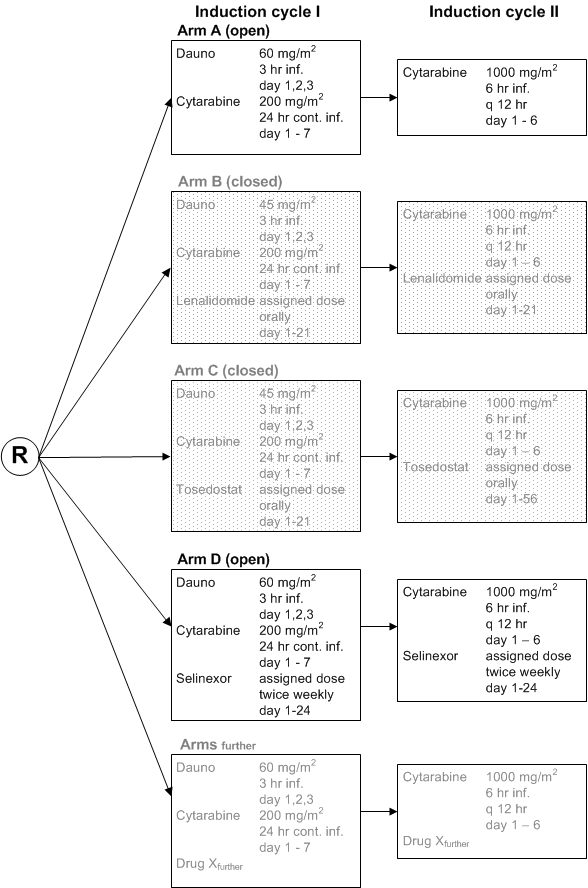
Details
- Phase:
- Prospective Phase III study
- Monitoring Type:
- Not any more
- Objectives:
Primary objectives
- For part A of the study (if applicable):
- 1. To assess the safety and tolerability of selinexor added to standard induction chemotherapy for AML (frequency and severity of toxicities and the durations of neutropenia and thrombocytopenia) and select the feasible dose level for part B
- 2. To assess in a randomized comparison the effect of selinexor on the CR rate.
- For part B:
- 1. To assess the safety and tolerability of selinexor added to standard induction chemotherapy for AML (frequency and severity of toxicities and the durations of neutropenia and thrombocytopenia) as regards the selected dose level of selinexor
- 2. To assess in a randomized comparison the effect of selinexor on the CR rate.
Secondary objectives
- For part B:
- 1. To determine the efficacy profile (event free survival (EFS) disease free survival (DFS) and overall survival (OS)) associated with the two therapy regimens.
- 2. To measure MRD by immunophenotyping in relation to clinical response parameters.
- 3. To identify potential biomarkers predictive of response, EFS, DFS and OS by exploratory genomic analysis (microarray, gene mutations)
- For part A of the study (if applicable):
Eligibility
- Inclusion Criteria:
- Patients eligible for standard chemotherapy.
- Patients 66 years and older
- Patients with a diagnosis of AML and related precursor neoplasms according to WHO 2008 classification (excluding acute promyelocytic leukemia) including secondary AML (after an antecedent hematological disease (e.g. MDS) and therapy-related AML, or acute leukemia’s of ambiguous lineage according to WHO 2008 or a diagnosis of refractory anemia with excess of blasts (MDS) and IPSS-R > 4.5
- Adequate renal and hepatic functions unless clearly disease related as indicated by the following laboratory values:
- Serum creatinine ≤1.0 mg/dL (≤88.7 µmol/L); if serum creatinine >1.0 mg/dL (>88.7 µmol/L), then the estimated glomerular filtration rate (GFR) must be >60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease equation where the Predicted GFR (ml/min/1.73 m2) = 186 x (Serum Creatinine in mg/dL)-1.154 x (age in years)-0.203 x (0.742 if patient is female) x (1.212 if patient is black) NOTE: if serum creatinine is measured in µmol/L, recalculate it in mg/dL according to the equation: 1 mg/dL = 88.7 µmol/L and use the above mentioned formula.
- Serum bilirubin ≤ 2.5 x upper limit of normal (ULN)
- Aspartate transaminase (AST) ≤ 2.5 x ULN
- Alanine transaminase (ALT) ≤ 2.5 x ULN
- Alkaline phosphatase ≤ 2.5 x ULN
- WHO performance status 0, 1 or 2 (see Appendix F)
- Written informed consent.
- Male and female patients must use an effective contraceptive method during the study and for a minimum of 6 months after study treatment.
- Exclusion Criteria:
- Acute promyelocytic leukemia
- Patients previously treated for AML (any antileukemic therapy including investigational agents), a short treatment period (< 2 weeks) with Hydroxyurea is allowed
- Concurrent history of active malignancy in the two past years prior to diagnosis except for:
- Basal and squamous cell carcinoma of the skin
- In situ carcinoma of the cervix
- Blast crisis of chronic myeloid leukemia
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, pulmonary disease etcetera)
- Cardiac dysfunction as defined by myocardial infarction within the last 6 months of study entry, or reduced left ventricular function with an ejection fraction < 50% ad measured by MUG scan or echocardiogram or unstable angina or New York Heart Association (NYHA) grade II or greater congestive heart failure (see Appendix I) or unstable cardiac arrhythmias
- Patients with a history of non-compliance to medical regimens or who are considered unreliable with respect to compliance
- Patients with any serious concomitant medical condition which could, in the opinion of the investigator, compromise participation in the study.
- Patients who have senile dementia, mental impairment or any other psychiatric disorder that prohibits the patient from understanding and giving informed consent.
- Current concomitant chemotherapy, radiation therapy, or immunotherapy other than as specified in the protocol.
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Eligible patients who have given written informed consent must be registered and randomised before start of treatment. For registration and randomization of patients we use our own eCRF, all patients are randomized between the standard treatment and one or more experimental treatments. Each hospital chooses to which set of experimental treatments it participates in the trial. So, all patients of an hospital are randomized over the same set of treatments, of course this set can change in time. To keep the number of patients who are treated with the standard treatment as small as possible we intend to randomize patients over more than one experimental treatment and the standard treatment.
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; Use goto eCRF button > select the [Patient tab] and click the [Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31 (0)10 704 1028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31 (0)10 704 1560 Monday through Friday, from 09:00 to 17:00 CET
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.