HOVON HO155 AML
Main info
- Identificatie:
- HOVON 155 AML
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Age:
- >= 18
- Stadium:
- 1st lijn
- Echelon:
- Limited Site Selection
- Included patients:
-
140(of 140)
- Active sites:
-
28(of 40)
- Title:
A randomized phase II multicenter study to assess the tolerability and efficacy of the addition of midostaurin to 10-day decitabine in unfit (i.e. HCT-CI ≥ 3) adult AML and high risk myelodysplasia (MDS) (IPSS-R > 4.5) patients.
A study in the frame of the masterprotocol of parallel randomized phase II studies in UNFIT- AML/high-risk MDS patients.
Timeline
News
16-11-2021: from 15Nov2021 the study is closed for inclusion, because the study has reached its target of 140 patients.
Patients who have started protocol treatment can continue their treatment according to protocol. The follow up will be according to protocol.
16-08-2021: The required number of 140 patients in de HO155 AML has almost been reached.
As of today only 10 patients can still be registered in HO155.
With the current inclusion rate we expect that the study will be closed for new patients within 1-2 months.
From now on, patient registration in ALEA will be exclusively done via the HOVON Data Center (HDC).
Participating sites can no longer register a patient in ALEA.
Important: “HOVON155 patient screening list” for potential patients
1. If you already had sent in screening samples of potential patients to the central lab, who are still wating to be registered, please inform us immediately
2. If you have a (new) potential patient for the HO155 study, please inform us first.
If there are still places available, your (potential)patient will be added to the “HOVON155 patient screening list”
By sending an email to: j.refos@erasmusmc.nl and hdc@erasmusmc.nl or Telephone +31 10 704 15 60
Please provide:
1. the year of birth
2. the planned date when is expected to register the patient in the HO155
Only patients listed on the “HO155 patient screening list” who are eligible will actually be registered by us.
08MAR2021: The following forms have been updated on the HOVON website:
- HO155 Dacogen drug order form_v4;
- HO155 Pharmacy info letter_v5 02MAR2021;
- HO155 AML Midostaurin_Shipping Form final_v2 MAR2021.
Flow
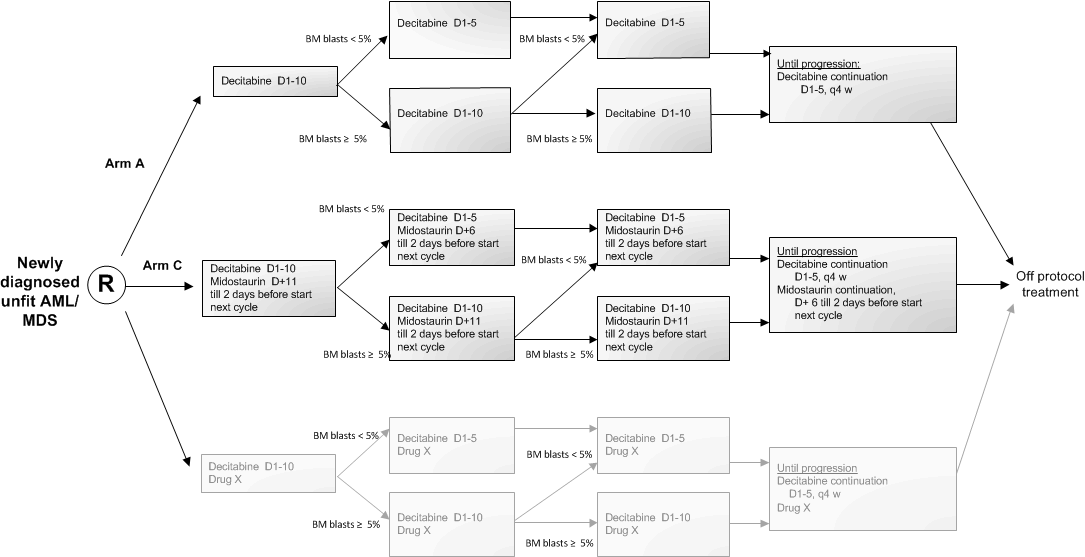
Details
- Phase:
- Prospective randomized Phase II study
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
Primary objectives
- To assess in a randomized comparison the effect of midostaurin added to 10-day decitabine treatment on the cumulative CR/CRi rate during 3 cycles.
Secondary objectives
- To assess the safety and tolerability of midostaurin added to 10-day decitabine treatment for AML (frequency and severity of toxicities and the durations of neutropenia and thrombocytopenia) as regards to the selected dose level of the study.
- To determine the efficacy profile: response rate (CRMRD-, CR, CRi, MLFS, PR), event free survival (EFS) and overall survival (OS) associated with the two therapy regimens.
- To measure MRD by immunophenotyping and PCR in relation to clinical response parameters.
- To identify gene mutations as potential biomarkers predictive of response, EFS and OS by exploratory analysis.
- To evaluate the prognostic value of baseline physical and functional conditions using comprehensive geriatric assessment tools (short physical performance battery (SPPB) and activities of daily living (ADL)) on treatment outcome.
Eligibility
- Inclusion Criteria:
- Patients with:
- a diagnosis of AML and related precursor neoplasms according to WHO 2016 classification (excluding acute promyelocytic leukemia) including secondary AML (after an antecedent hematological disease (e.g. MDS) and therapy-related AML, or
- a diagnosis of myelodysplastic syndrome with excess of blasts (MDS) and IPSS-R > 4.5
- Patients 18 years and older.
- Patients NOT eligible for standard chemotherapy, defined as HCT-CI ≥ 3. (Appendix G) or Patients NOT eligible for standard chemotherapy for other reasons (wish of patient).
- WBC ≤ 30 x10^9/L (prior hydroxyurea allowed for a maximum of 5 days, stop 2 days before start decitabine treatment)
- Adequate renal and hepatic functions unless clearly disease related as indicated by the following laboratory values:
- Serum creatinine ≤ 221.7 μmol/L (≤ 2.5 mg/dL ), unless considered AML-related
- Serum bilirubin ≤ 2.5 x upper limit of normal (ULN), unless considered AML-related or due to Gilbert’s syndrome
- Alanine transaminase (ALT) ≤ 2.5 x ULN, unless considered AML-related
- WHO performance status 0, 1 or 2 (see Appendix D).
- Patient is willing and able to use adequate contraception during and until 5 months after the last protocol treatment.
- Written informed consent.
- Patient is capable of giving informed consent.
- Patients with:
- Exclusion Criteria:
- Acute promyelocytic leukemia.
- Acute leukemia's of ambiguous lineage according to WHO 2016
- Patient has symptomatic central nervous system (CNS) leukemia (NO routinely lumbar puncture required to investigate CNS involvement)
- Blast crisis of chronic myeloid leukemia.
- Diagnosis of any previous or concomitant malignancy is an exclusion criterion:
- except when the patient completed successfully treatment (chemotherapy and/or surgery and/or radiotherapy) with curative intent for this malignancy at least 6 months prior to randomization. OR
- except for basal and squamous cell carcinoma of the skin or in situ carcinoma of the cervix
- Patients previously treated for AML (any antileukemic therapy including investigational agents), a short treatment period ( ≤ 5 days) with Hydroxyurea is allowed
- Current concomitant chemotherapy, radiation therapy, or immunotherapy; other than hydroxyurea
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled diabetes, infection, hypertension, pulmonary disease etc.)
- Cardiac dysfunction as defined by:
- Myocardial infarction within the last 3 months of study entry, or
- Reduced left ventricular function with an ejection fraction < 40% as measured by MUGA scan or echocardiogram or
- Unstable angina or
- New York Heart Association (NYHA) grade IV congestive heart failure (see Appendix I) or
- Unstable cardiac arrhythmias.
- History of stroke or intracranial hemorrhage within 6 months prior to randomization.
- Patient has a history of human immunodeficiency virus (HIV) or active infection with Hepatitis C or B.
- Patients known to be pregnant
- Patients with a history of non-compliance to medical regimens or who are considered unreliable with respect to compliance.
- Patients with any serious concomitant medical condition which could, in the opinion of the investigator, compromise participation in the study.
- Patients who have senile dementia, mental impairment or any other psychiatric disorder that prohibits the patient from understanding and giving informed consent.
- Any psychological, familial, sociological or geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- HOVON Data Center registration database (www.hovon.nl). Account for registration and a registration manual can be requested at HOVON Data Center.
- By sending the completed registration/randomization CRF by email to hdc@erasmusmc.nl, Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Sex
- Year of birth,
- Age at date of inclusion
- Date written informed consent
- Specific items patient gives consent for (see ICF)
- Eligibility criteria
- Stratification factors
All eligibility criteria will be checked with a checklist.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.