HOVON HO159 CLL
Open
Main info
- Identificatie:
- HO159 CLL
- Sponsor:
- HOVON
- Working group party:
- CLL
- Age:
- >= 18
- Stadium:
- 2de lijn
- Echelon:
- Level D
- Included patients:
-
51(of 60)
- Active sites:
-
20(of 20)
- Title:
A prospective, multicenter, phase-II trial of venetoclax plus acalabrutinib in patients who have relapsed after first line venetoclax + anti-CD20 mAb treatment for chronic lymphocytic leukemia (CLL or SLL)
Timeline
Scheduled
Actual
2019
01 mei
Development
2020
14 apr.
Submission in Progress
2020
01 mei
EC Approval
2020
23 jul.
EC Approval
2020
15 dec.
Activated
2020
15 dec.
First Site
2020
23 dec.
First Site
2020
23 dec.
Activated
2020
31 dec.
FPI
2021
03 jun.
FPI
2026
31 mrt.
ClosedForInclusionScheduledStart
2029
18 jun.
Endpoint Analysis
2032
31 mei
Closeout in Progress
2033
31 mei
Archived
News
23MAR2026: An update of the contactsheet is available on the website.
Flow
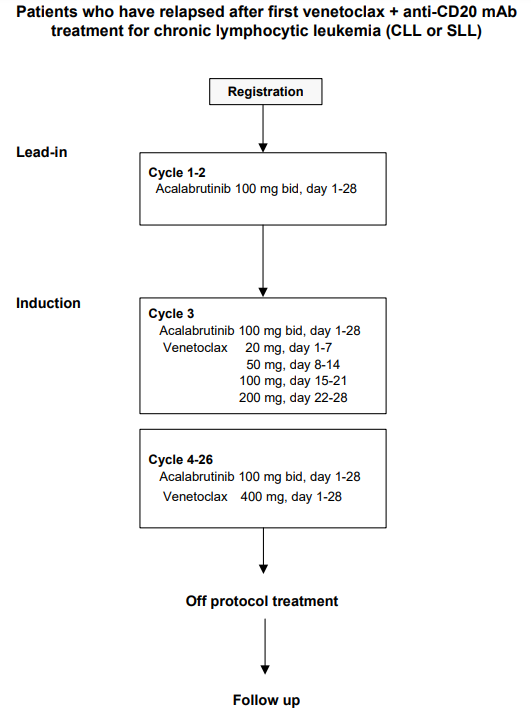
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
Primary Objective
- To evaluate efficacy of acalabrutinib/venetoclax (AV) in terms of uMRD response in BM after 26 cycles of treatment in patients with CLL previously treated with venetoclax and anti-CD20 mAb.
Secondary Objectives
- To evaluate the efficacy of AV.
- To evaluate the safety and tolerability of AV.
Exploratory
- To evaluate prognostic parameters for efficacy
- To evaluate value of different techniques for MRD testing.
- To evaluate the impact on immunological function of AV.
- To evaluate grading for hematological toxicity according to International Workshop on Chronic Lymphocytic Leukemia (IWCLL)19 by assessing lab values.
- To evaluate quality of life (QoL) with AV.
- To asses impact on venetoclax pharmacokinetics (PK) in combination with acalabrutinib
Eligibility
- Inclusion Criteria:
- Documented CLL or SLL requiring treatment according to IWCLL criteria (appendix A) after at least (clinical) partial response as best response after the following initial study treatment: venetoclax-rituximab in HOVON 140/GAIA or venetoclax-obinutuzumab in HOVON 139/GIVE or HOVON 140/GAIA;
- WHO/ECOG performance status 0-3 (appendix C), stage 3 only if attributable to CLL
- Age at least 18 years;
- Adequate BM function defined as:
- Hemoglobin >5 mmol/l or Hb > 8 g/dL
- Absolute neutrophil count (ANC) >0.75 x 109/L (750/μL), unless directly attributable to CLL infiltration of the BM, proven by BM biopsy
- Platelet count >30 x 109/L (30,000/μL) without transfusion and irrespective whether it is attributable to CLL infiltration in the BM;
- Estimated Glomerular Filtration Rate (eGFR) (MDRD) or estimated creatinine clearance (CrCl ≥ 30ml/min (Cockcroft-Gault appendix E); Please note: in case eGFR or CrCl is <50ml/min the patient needs to be considered high risk for TLS
- Adequate liver function as indicated:
- Serum aspartate transaminase (ASAT) and alanine transaminase (ALAT) ≤ 3.0 x upper limit of normal (ULN);
- Bilirubin ≤1.5 x ULN (unless bilirubin rise is due to Gilbert's syndrome or of nonhepatic origin);
- Prothrombin time (PT)/International normal ratio (INR) <1.5 x ULN and activated partial thromboplastin time (aPTT) <1.5 x ULN;
- Negative serological testing for hepatitis B virus (HBV) (Hepatitis B surface antigen (HBsAg) negative and hepatitis B core antibody (anti-HBc) negative) and hepatitis C virus (hepatitis C antibody). Subjects who are positive for anti-HBc or hepatitis C antibody may be included if they have a negative PCR within 6 weeks before enrollment. Those who are PCR positive will be excluded; Please note: For patients positive for anti-HBc HBV-DNA PCR has to be repeated every month until 12 months after last dose of study treatment.
- Patient is able and willing to adhere to the study visit schedule and other protocol requirements;
- Patient is capable of giving informed consent;
- Written informed consent.
- Exclusion Criteria:
- Any prior therapy with BTK inhibitor;
- Prior treatment with venetoclax other than first line;
- Other therapy with exception of chemo-/immunotherapy which is allowed also after venetoclax first line relapse;
- Transformation of CLL (Richter’s transformation);
- Patient with a history of confirmed progressive multifocal leukoencephalopathy (PML);
- Malignancies other than CLL currently requiring systemic therapy or not treated in curative intention or showing signs of progression after curative treatment;
- Known allergy to xanthine oxidase inhibitors and/or rasburicase;
- History of drug-specific hypersensitivity or anaphylaxis to any study drug (including active product or excipient components);
- Active bleeding or history of bleeding diathesis (e.g. hemophilia or von Willebrand disease);
- Active fungal, bacterial, and/or viral infection that requires systemic therapy; Please note: active controlled as well as chronic/recurrent infections are at risk of reactivation/infection during treatment;
- Concurrent severe and/or uncontrolled medical condition (e.g. uncontrolled: infection, auto-immune hemolysis, immune thrombocytopenia, diabetes, hypertension, hyperthyroidism or hypothyroidism etc.);
- Patient known to be HIV-positive;
- Patient requiring treatment with a strong cytochrome P450 (CYP) 3A inhibitor/inducer (see appendix J) or anticoagulant therapy with warfarin or phenoprocoumon or other vitamin K antagonists; Please note: Patients being treated with DOACs apixaban, edoxaban or rivaroxaban can be included, but must be properly informed about the potential risk of bleeding under treatment with acalabrutinib. (see appendix J)
- History of stroke or intracranial hemorrhage within 6 months prior to registration;
- Severe cardiovascular disease (arrhythmias requiring chronic treatment, congestive heart failure or symptomatic ischemic heart disease, myocardial infarction within 6 months) (CTCAE grade III-IV, see appendix D);
- Severe pulmonary dysfunction (CTCAE grade III-IV, see appendix D);
- Severe neurological or psychiatric disease (CTCAE grade III-IV, see appendix D);
- Patient who has difficulty with or are unable to swallow oral medication, or have significant gastrointestinal disease that would limit absorption of oral medication;
- Vaccination with live vaccines within 28 days prior to registration;
- Use of any other experimental drug or therapy within 28 days of registration;
- Major surgery within 28 days prior to registration;
- Steroid therapy within 10 days prior to registration, with the exception of inhaled steroids for asthma, topical steroids, steroids up to 20 mg or dose equivalents of prednisolone daily to control autoimmune phenomenon’s, or replacement/stress corticosteroids;
- Pregnant women and nursing mothers;
- Fertile men or women of childbearing potential unless: (1) surgically sterile or ≥ 2 years after the onset of menopause; (2) willing to use a highly effective contraceptive method such as oral contraceptives, intrauterine device, sexual abstinence or combination of male condom with either cap, diaphragm, or sponge with spermicide (double barrier methods) during study treatment and for 30 days after end of treatment;
- Current participation in other clinical trial (other than follow up HOVON139/HOVON140);
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; select the [patient] tab and click the [ Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31.10.7041028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
When the first 5 patients have finished cycle 4.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.
Site
20 results
Order by
Accrual rate
Activation date
NL-Rotterdam-MAASSTADZIEKENHUIS
6
12 apr. 2021
NL-Arnhem-RIJNSTATE
4
04 jun. 2021
NL-Leeuwarden-MCL
4
23 dec. 2020
NL-Groningen-UMCG
4
18 mrt. 2021
NL-Ede-ZGV
4
24 dec. 2020
NL-Delft-RDGG
4
10 mei 2021
NL-Breda-AMPHIA
4
11 jan. 2021
NL-Amsterdam-AMC
4
23 dec. 2020
NL-Rotterdam-IKAZIA
3
15 jul. 2021
NL-Utrecht-UMCUTRECHT
3
04 mei 2022
NL-Nieuwegein-ANTONIUS
3
14 jan. 2021
NL-Den Bosch-JBZ
2
23 dec. 2020
NL-Maastricht-MUMC
1
15 feb. 2021
DK-Aarhus N-AUH
1
11 sep. 2023
NL-Dordrecht-ASZ
1
06 sep. 2022
BE-Bruxelles-STLUC
1
09 nov. 2021
NL-Eindhoven-MAXIMAMC
1
19 jan. 2021
NL-Terneuzen-ZORGSAAM
1
20 jan. 2023
NL-Gouda-GROENEHART
22 dec. 2022
BE-Leuven-UZLEUVEN
09 nov. 2021