HOVON HO114 MM
Main info
- Identificatie:
- HOVON 114 MM / EMN11
- Sponsor:
- HOVON
- Working group party:
- Myeloma
- Age:
- >= 18
- Stadium:
- 2de lijn
- Echelon:
- Limited Site Selection
- Included patients:
-
112(of 222)
- Active sites:
-
33(of 50)
- Title:
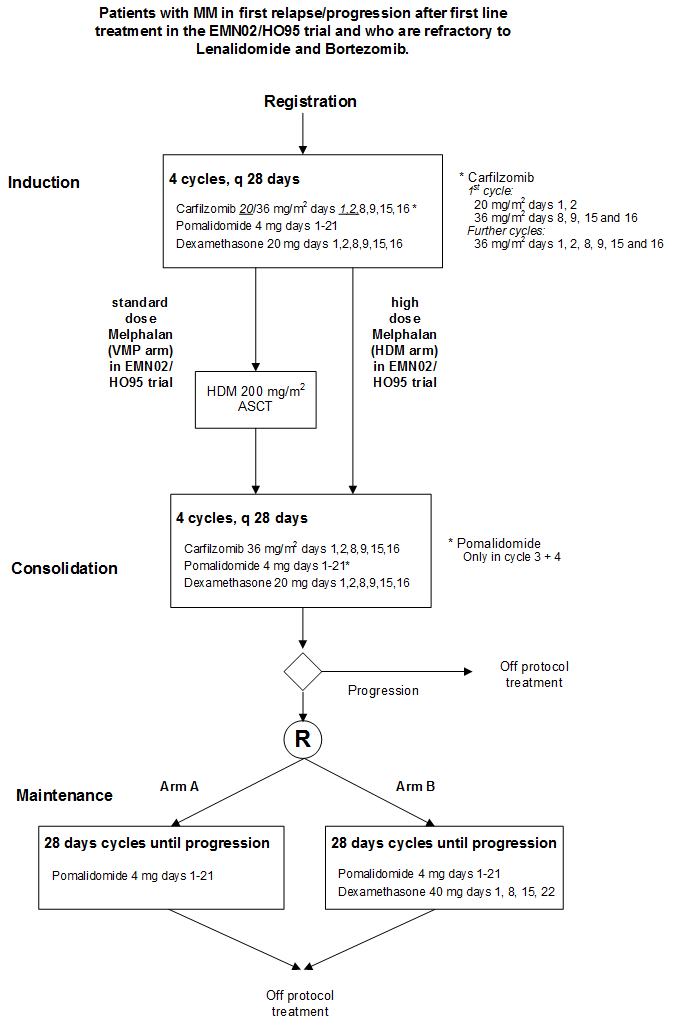
Pomalidomide combined with Carfilzomib and Dexamethasone (PCd) for induction and consolidation followed by Pomalidomide combined with Dexamethason vs Pomalidomide maintenance for patients with Multiple Myeloma in progression after prior 1st line treatment with Lenalidomide and Bortezomib.
Timeline
News
Addendum ICF to inform the patients about the study stop in 2024 is available:
HOVON will end the HO114/EMN11 study as of July 2024. Continuous treatment with Pomalidomide has already been included in the treatment guideline for Multiple Myeloma patients.
Flow
Details
- Phase:
- Prospective randomized Phase II study
- Monitoring Type:
- Not any more
- Objectives:
Primary objectives
- Evaluate the efficacy, defined as PFS, of pomalidomide maintenance plus dexamethasone versus pomalidomide maintenance in patients who responded (≥ PR) to the combination of pomalidomide (POM), carfilzomib (CAR) and low dose dexamethasone (LD-DEX) for induction and consolidation .
- Evaluate efficacy defined as response rate (sCR, CR, VGPR, PR) of the combination of pomalidomide (POM), carfilzomib (CAR) and low dose dexamethasone (LD-DEX) for induction and consolidation in subjects with relapsed or refractory multiple myeloma (MM) after prior first-line treatment in the EMN02/HO95 trial who are refractory to Lenalidomide and/or Bortezomib. This objective will be investigated in patients who have or have not received a prior autologous transplant.
Secondary objectives
- Evaluate the response rate after 8 cycles of PCd before the start of maintenance.
- Evaluate the safety and tolerability of the combination of pomalidomide, carfilzomib and low dose dexamethasone in subjects with relapsed or refractory multiple myeloma.
Exploratory objectives
- Evaluation of biomarkers, including baseline markers predictive of response to pomalidomide combined with carfilzomib and dexamethasone
- Evaluate the quality of life
- Evaluate the gene expression profiles and SNPs related to treatment outcome and side effects
Eligibility
- Inclusion Criteria:
- Included in EMN02/HO95 trial. Induction therapy followed by autologous stem cell transplant (AutoSCT) and consolidation/ maintenance will be considered as one regimen.
- The subject must understand and voluntarily sign an informed consent document prior to any study related assessments/procedures.
- Age ≥ 18 years at the time of signing the informed consent form.
- Able to adhere to the study visit schedule and other protocol requirements.
- Documented diagnosis of multiple myeloma and measurable disease (serum M-protein ≥ 10 g/L or urine M-protein ≥ 200 mg/24 hours or abnormal FLC ratio with involved free light chain (FLC) > 100 mg/L) or proven plasmacytoma by biopsy).
- Documented progression or refractory multiple myeloma as per the IMWG uniform response criteria (Durie, 2006) during or after the EMN02/HO95 trial. Normal renal function with a Creatinine Clearance > 45mL/min according to the Modification of Diet in Renal Disease (MDRD) equation for estimation of Glomerular Filtration Rate (GFR)
- WHO performance status score of 0, 1 or 2.
- Patients must be willing and capable to use adequate contraception during the therapy (all men, all pre-menopausal women).
- Patients must be able to adhere to the requirements of the Pregnancy Prevention Risk Management Plan.
- Patients must be eligible for autologous stem cell transplantation when not previously given in first line treatment.
- All subjects must agree to refrain from donating blood while on study drug and for 28 days after discontinuation from this study treatment.
- All subjects must agree not to share medication.
- Exclusion Criteria:
- Patient received more than 1 regimen (EMN02/HO95), except local radiotherapy.
- Absolute neutrophil count (ANC) <1.0 x 10^9/L, unless related to MM.
- Platelet count < 75 x 10^9/L, unless related to MM.
- Corrected serum calcium > 14 mg/dL (> 3.5 mmol/L).
- Hemoglobin < 8 g/dL (< 4.9 mmol/L; prior RBC transfusion or recombinant human erythropoietin use is permitted).
- Significant hepatic dysfunction (Serum SGOT/AST or SGPT/ALT > 3.0 x upper limit of normal (ULN) or serum total bilirubin > 3.0 x ULN)
- Prior history of malignancies, other than MM, unless the subject has been free of the disease for ≥ 5 years. Exceptions include the following:
- Basal or squamous cell carcinoma of the skin.
- Carcinoma in situ of the cervix or breast.
- Incidental histological finding of prostate cancer (TNM stage of T1a or T1b).
- Previous therapy with pomalidomide or carfilzomib.
- Hypersensitivity to thalidomide, lenalidomide, bortezomib or dexamethasone (this includes ≥ Grade 3 rash during prior thalidomide or lenalidomide or bortezomib therapy).
- Peripheral neuropathy ≥ Grade 2.
- Subjects who received an allogeneic bone marrow or allogeneic peripheral blood stem cell transplant less than 12 months prior to initiation of study treatment.
- LVEF ≤ 40%.
- QTc > 450 msec.
- History of torsade de pointes.
- History of ventricular tachycardia, ventricular fibrillation.
- Uncontrolled atrial fibrillation/flatter.
- Congestive heart failure (NY Heart Association Class III or IV).
- Myocardial infarction within 12 months prior to starting study treatment
- Unstable or poorly controlled angina pectoris, including Prinzmetal variant angina pectoris.
- History of pulmonary hypertension.
- Uncontrolled infection.
- Subjects who received any of the following within the last 14 days of initiation of study treatment:
- Major surgery (kyphoplasty is not considered major surgery).
- Use of any anti-myeloma drug therapy.
- Use of any investigational agents (with the exception of lenalidomide) within 28 days or five half-lives (whichever is longer) of treatment.
- Incidence of gastrointestinal disease that may significantly alter the absorption of pomalidomide.
- Subjects unable or unwilling to undergo antithrombotic prophylactic treatment.
- Any serious medical condition, laboratory abnormality, or psychiatric illness that would prevent the subjects from signing the informed consent form.
- Pregnant or breastfeeding females.
- Known human immunodeficiency virus (HIV) positivity, active infectious hepatitis A, B or C or chronic hepatitis B or C.
- Pre-existing pulmonary, cardiac or renal impairement that prevents hydration measures as described at section 9.5.
- Any psychological, familial, sociological and geographical condition potentially hampering compliance with the study protocol and follow-up schedule.
Registration Details
Eligible patients should be registered before start of treatment. Patients will be registered at the EMN Data Center by web http://www.emntg.org. Investigators who do not have an account yet should register at this website to obtain an account after consultation of HDC.
The following information will be requested at registration:
- Protocol number
- Institution name
- Name of caller/responsible investigator
- Sex
- Date of birth
- Date written informed consent
- Specific items patient gives consent for (see ICF)
- Eligibility criteria
- Criteria for measurable disease and CRAB criteria
All eligibility criteria will be checked with a checklist.
Each patient will be given a unique patient study number
One interim analysis is planned, primarily to describe adverse events observed during the carfilzomib + pomalidomide + dexamethasone re-induction chemotherapy. This will be done when data of the first 20 patients completing the 4 cycles of induction therapy are available.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.