HOVON HO156 AML
Main info
- Identificatie:
- HOVON 156 AML_AMLSG 28-18
- Sponsor:
- HOVON
- Working group party:
- Leukemia
- Stadium:
- 1st lijn
- Echelon:
- Level C-HIC&C-SCT
- Included patients:
-
777(of 777)
- Active sites:
-
190(of 200)
- Title:
A phase 3, multicenter, open-label, randomized, study of Gilteritinib versus Midostaurin in combination with induction and consolidation therapy followed by one-year maintenance in patients with newly diagnosed Acute Myeloid Leukemia (AML) or Myelodysplastic syndromes with excess blasts-2 (MDS-EB2) with FLT3 mutations eligible for intensive chemotherapy.
Timeline
News
16APR2024 We have updated the eCRF instructions and also included the new version of the protocol (v07)
13JUN2023 We have reached the LPI! We thank you all for your help in reaching this milestone!
15MAY2023 Registration reactivated in most other countries as well
For Belgium we now also receveid approval, therefore these sites will be activated as well. Also in FR, DE, CH the trial has been reactivated
27MAR2023 Registration activated!
For the Netherlands we have receveid approval, therefore the trial was activated for the Dutch sites. We hope that the other countries will follow soon!
11JAN2023 New SAE form
As it was sometimes hard to read the SAE forms that were send in, we have converted the SAE form in a fillable format, so that it also can be filled out electronically.
09DEC2022 Inclusion placed on hold
Please note that on advice of the DSMB the inclusion has been placed on hold for the time being. Participating sites will be informed by email.
05DEC2022 New eCRF instructions
A new version of the eCRF instructions is availabele.
10MAR2022 New lab manual
The new lab manual has been sent to all paricipating sites.
02MAR2022 New pharmacy manual
The new pharmacy manual has been sent to all paricipating sites.
03JAN2022 Amendment 08 role out - BE
For the Netherlands the approvals have been received and the documents have been shared to all participating sites by email concerning the new amendment.
13OCT2021 Amendment 08 role out - NL
For the Netherlands the approvals have been received and the documents have been shared to all participating sites by email concerning the new amendment.
19MAY2021 Correction on the EC approval
With regards to the EC approval, in their list of approved documents the EC had by accident not copied down the correct IB midostaurin version 24 date. This was mentioned as 24-7-2019. They have now updated it to the correct date: 2-4-2020. The updated EC aproval can be found in the downloads section.
19MAY2021 Usage of Addendum
For the Netherlands we received the question if the Addendum should be used for all patients, or just the Arm B (Gilteritinib) patients, as the updates are only relevant for them. We have requested this to our central EC, and indeed they approve that only patients that signed the previous ICF (02APR2019) and were randomized to Arm B need to sign the addendum.
16DEC2020 Update site lab manual
We have send out the site labmanual to all active sites, this document can also be found in the study documents at the bottom of this page.
04MAR2020 IF update
The IF has been updated to add a very small update of the assessment instructions that incorrectly indicated page 1 of 2 in to the top right corner, while it only existed out of 1 page. As this update is so minor it was not send out to the sites separately.
14JAN2020 IF update
The IF has been update to add the latest version of the lab manual.
20DEC2019 First study site activation
Very first study site (NL-Rotterdam-ERASMUSMC) has been activated.
02DEC2019 IF update
The IF has been update to add the eCRF instructions as well.
27NOV2019 IF update
The IF has been updated to add the new pharmacy manual.
18NOV2019 Study activation
Study has been activated
01NOV2019 IF update
The ITF has been updated as we received a new IB for Midosaturin (v23). We have updated the IF accordingly. Please download the latest IF. Also the site document checklist has been updated as the CV of the independent physician is also needed for site activation.
30OCT2019 IF update
The IF has been updated as we received a new QP declaration and a new IB for Gilteritinib. We have updated the IF accordingly. Please download the latest IF.
12SEP2019 (Investigator File uploaded)
All Dutch sites have received by email a link to the IF that contains all documents required to receive local approval. This IF contains documents like: Protocol, ICFs (both study ICF and screenings ICF), ABR form, EC correspondence and Central EC approval form, CA approval, Patient card (which you will also receive by regular post), Patient Diaries (for both Midostaurin and Gilteritinib), eCRF instructions, SAE forms, etc.
Below - in section Downloads you can download the entire zip file as well. You can also find an excel sheet (HO156_Overview folders and files_ddMONyyyy) that lists the exact documents that are within the ZIP file and indicate the section in which they are filed. If there are updates, these will be emailed to the sites, the latest version of the ZIP file can always be downloaded below.
Flow
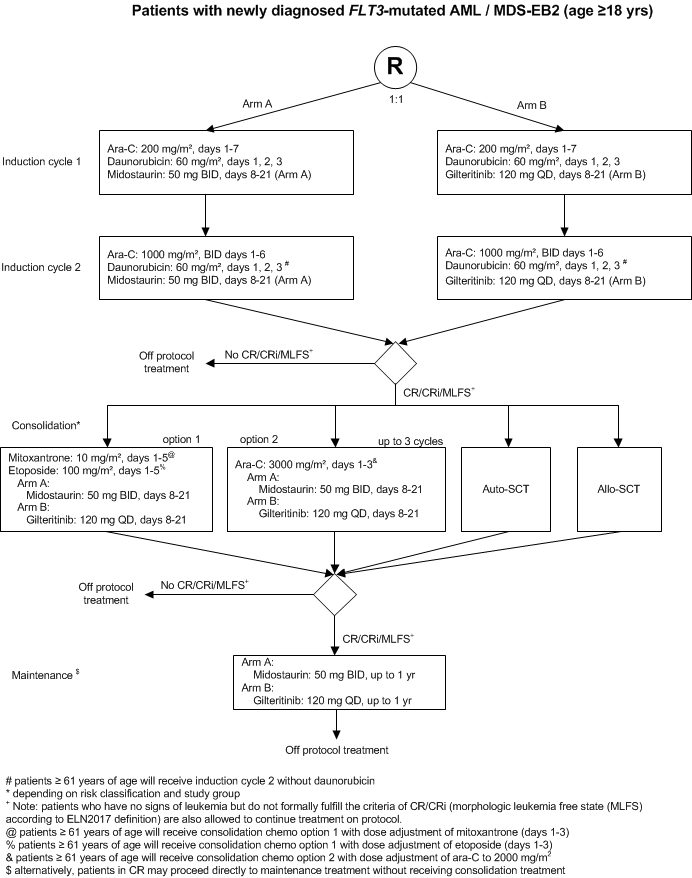
Details
- Phase:
- Prospective randomized Phase III study
- Monitoring Type:
- Study Specific
- Objectives:
Primary study objective
- To compare OS between gilteritinib and midostaurin in combination with induction therapy and consolidation therapy followed by one-year maintenance therapy in patients with newly diagnosed AML with a FLT3 gene mutation eligible for intensive chemotherapy.
Key secondary study objectives
- To determine if treatment with gilteritinib, as compared to midostaurin, prolongs EFS in the AML patient group.
- To compare the complete remission (CR) rate after induction therapy (i.e., CR as best response during or at completion of induction) for treatment including gilteritinib vs. midostaurin in the AML patient group.
- To determine if treatment with gilteritinib, as compared to midostaurin, prolongs EFS with a modified CR by 60 days after initiation of the last induction cycle (mEFS) in the AML patient group.
Other secondary study objectives
- To compare CR and CR with incomplete hematologic recovery (CRi) rates after induction cycle 1 and after induction cycle 2 for treatment including gilteritinib vs. midostaurin in the AML patient group.
- To compare relapse-free survival (RFS), cumulative incidence of relapse (CIR) and death (CID) for treatment including gilteritinib vs. midostaurin in the AML patient group.
- To evaluate minimal residual disease (MRD) status at sequential time points throughout treatment and CRMRD− and CR/CRiMRD- rates between treatment including gilteritinib vs. midostaurin, using molecular and/or flow cytometric techniques in the AML patient group.
- To assess the safety and tolerability of treatment including gilteritinib vs. midostaurin in the AML patient group.
- To assess the time to hematopoietic recovery (ANC 0.5 and 1.0x109/L; platelets 50 and 100x109/L) after each chemotherapy treatment cycle in the AML patient group.
- To assess the percentage of patients undergoing an allogeneic stem cell transplant (allo-SCT) in the AML patient group.
- To determine quality of life (QoL) during maintenance treatment with gilteritinib vs. midostaurin in the AML patient group.
Exporatory objectives
- To evaluate the above mentioned endpoints between gilteritinib and midostaurin in combination with induction and consolidation therapy followed by one-year maintenance therapy in patients with Myelodysplastic Syndromes (MDS) with excess blasts-2 (EB2) with a FLT3 gene mutation eligible for intensive chemotherapy
Eligibility
- Inclusion Criteria:
- Age ≥18 years
- Newly diagnosed AML or MDS with excess of blasts-2 (EB2) defined according to WHO criteria (appendix A), with centrally documented FLT3 gene mutation (either TKD or ITD or both). AML may be secondary to prior hematological disorders, including MDS, and/or therapy-related. Patients may have had previous treatment with erythropoiesis stimulating agents (ESA) and/or hypomethylating agents (HMAs) for MDS. ESA and HMAs have to be stopped at least four weeks before registration
- FLT3 mutation as assessed by DNA fragment analysis PCR for FLT3-ITD and sequencing for FLT3-TKD. FLT3-ITD positivity is defined as a FLT3-ITD / FLT3-WT ratio of ≥ 0.05 (5%). FLT3-TKD positivity is defined as a variant allelele frequency (VAF = FLT3 TKD / (FLT3 WT + FLT3 TKD)) ≥ 0.05 (5%))
- Considered to be eligible for intensive chemotherapy
- Patient is suitable for oral administration of study drug
- WHO/ECOG performance status ≤ 2
- Adequate hepatic function as evidenced by
- Serum total bilirubin ≤ 2.5 × upper limit of normal (ULN) unless considered due to leukemic involvement following written approval by the (co) Principal Investigator
- Aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP) ≤ 3.0 × ULN, unless considered due to leukemic involvement following written approval by the (co) Principal Investigator
- Adequate renal function as defined by creatinine clearance > 40 mL/min based on the Cockroft-Gault glomerular filtration rate (GFR)
- Written informed consent
- Patient is capable of giving informed consent
- Female patient must either:
- Be of nonchildbearing potential:
- Postmenopausal (defined as at least 1 year without any menses) prior to screening, or
- Documented surgically sterile or status posthysterectomy (at least 1 month prior to screening)
- Or, if of childbearing potential,
- Agree not to try to become pregnant during the study and for 6 months after the final study drug administration
- And have a negative urine or serum pregnancy test at screening
- And, if heterosexually active, agree to consistently use highly effective contraception per locally accepted standards in addition to a barrier method starting at screening and throughout the study period and for 6 months after the final study drug administration.
- Highly effective forms of birth control include:
- Consistent and correct usage of established hormonal contraceptives that inhibit ovulation,
- Established intrauterine device (IUD) or intrauterine system (IUS),
- Bilateral tubal occlusion,
- Vasectomy (A vasectomy is a highly effective contraception method provided the absence of sperm has been confirmed. If not, an additional highly effective method of contraception should be used.)
- Male is sterile due to a bilateral orchiectomy.
- Sexual abstinence is considered a highly effective method only if defined as refraining from heterosexual activity during the entire period of risk associated with the study drug. The reliability of sexual abstinence needs to be evaluated in relation to the duration of the clinical study and the preferred and usual lifestyle of the patient.
- List is not all inclusive. Prior to enrollment, the investigator is responsible for confirming patient will utilize highly effective forms of birth control per locally accepted standards during the protocol defined period.
- Female patient must agree not to breastfeed starting at screening and throughout the study period, and for 2 months and 1 week after the final study drug administration.
- Female patient must not donate ova starting at screening and throughout the study period, and for 6 months after the final study drug administration.
- Male patient and their female partners who are of childbearing potential must be using highly effective contraception per locally accepted standards in addition to a barrier method starting at screening and continue throughout the study period and for 4 months and 1 week after the final study drug administration.
- Male patient must not donate sperm starting at screening and throughout the study period and for 4 months and 1 week after the final study drug administration.
- Patient agrees not to participate in another interventional study while on treatment
- Exclusion Criteria:
- Prior chemotherapy for AML or MDS-EB2 (with the exception of ESA and HMA). Hydroxyurea is allowed for the control of peripheral leukemic blasts in patients with leukocytosis (e.g., white blood cell [WBC] counts > 30 x 109/L)
- Acute promyelocytic leukemia (APL) with PML-RARA or one of the other pathognomonic variant fusion genes/chromosome translocations
- Blast crisis after CML
- Patient requires treatment with concomitant drugs that are strong inducers of cytochrome P450 (CYP) 3A (Appendix I)
- Breast feeding as of the start of study treatment
- Active infection, including hepatitis B or C or HIV infection that is uncontrolled at randomization. An infection controlled with an approved or closely monitored antibiotic/antiviral/antifungal treatment is allowed.
- Patients with a currently active second malignancy. Patients are not considered to have a currently active malignancy if they have completed therapy and are considered by their physician to be at less than 30% risk of relapse within one year. However, patients with the following history/concurrent conditions are allowed:
- Basal or squamous cell carcinoma of the skin;
- Carcinoma in situ of the cervix;
- Carcinoma in situ of the breast;
- Incidental histologic finding of prostate cancer
- Significant active cardiac disease within 6 months prior to the start of study treatment, including:
- New York Heart Association (NYHA) Class III or IV congestive heart failure;
- Myocardial infarction;
- Unstable angina and/or stroke;
- Left ventricular ejection fraction (LVEF) < 40% by ECHO or MUGA scan obtained within 28 days prior to the start of study treatment
- QTc interval using Fridericia’s formula (QTcF) ≥ 450 msec (average of triplicate determinations) or other factors that increase the risk of QT prolongation or arrhythmic events (e.g., heart failure, family history of long QT interval syndrome). Prolonged QTc interval associated with bundle branch block or pacemaking is permitted with written approval of the (co) Principal Investigator.
- Patient with hypokalemia and hypomagnesemia at screening (defined as values below LLN)
- Dysphagia, short-gut syndrome, gastroparesis, or other conditions that limit the ingestion or gastrointestinal absorption of orally administered drugs
- Clinical symptoms suggestive of active central nervous system (CNS) leukemia or known CNS leukemia. Evaluation of cerebrospinal fluid (CSF) during screening is only required if there is a clinical suspicion of CNS involvement by leukemia during screening
- Immediate life-threatening, severe complications of leukemia such as uncontrolled bleeding and/or disseminated intravascular coagulation
- Any other medical or psychological condition deemed by the Investigator to be likely to interfere with a patient’s ability to give informed consent or participate in the study
- Any psychological, familial, sociological or geographical condition potentially hampering compliance with the study protocol and follow-up schedule
Registration Details
Patients who are possibly eligible for this trial should first be screened to determine the FLT3 mutation status. This can only be done after the patient has provided a written informed consent for the screening. Screening is done through a separate screening database. There are two screening databases for this trial, one managed by HOVON and one managed by AMLSG. Each group/institute that is participating in this trial is assigned one of the two screening databases.
Eligible patients should be registered before start of treatment. Patients need to be registered at the HOVON Data Center by one of the following options:
- By ALEA; Use goto eCRF button > select the [Patient tab] and click the [Add new patient] button. Complete all items and click the [Submit] button
- By faxing the completed registration/randomization CRF +31 (0)10 704 1028 Monday through Friday, from 09:00 to 17:00 CET
- By phone +31 (0)10 704 1560 Monday through Friday, from 09:00 to 17:00 CET
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.