HOVON HO169 NHL
Main info
- Identificatie:
- HO169 CAD
- Sponsor:
- HOVON
- Working group party:
- Lymphoma
- Age:
- >= 18
- Stadium:
- Alle lijnen
- Echelon:
- Level A
- Included patients:
-
26(of 26)
- Active sites:
-
9(of 10)2 sites are pending
- Title:
HOVON 169 CAD: Phase II trial of Zanubrutinib in primary Cold Agglutinin Disease.
CaZa study
Timeline
Flow
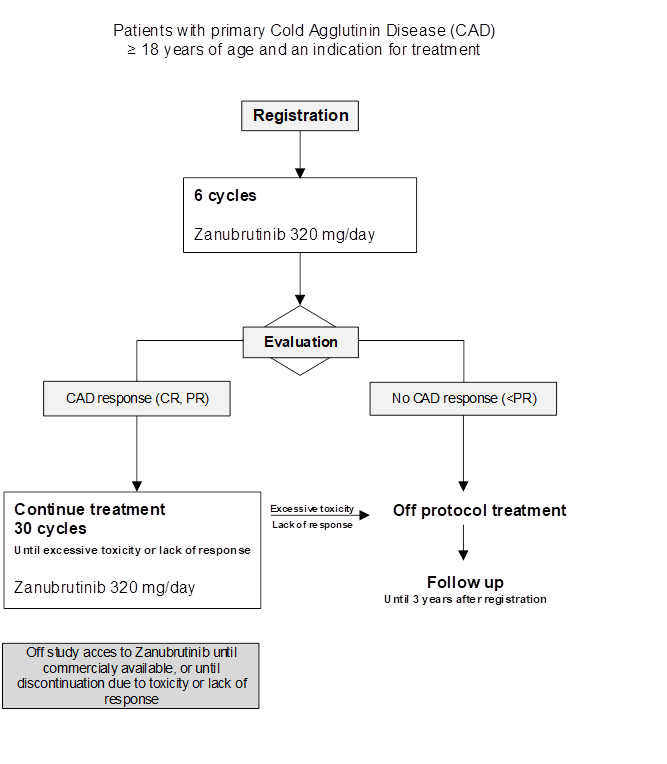
Details
- Phase:
- Prospective Phase II study
- Monitoring Type:
- Site Evaluation Visit
- Objectives:
To evaluate the efficacy of 6 cycles of zanubrutinib (based on CAD response) in patient
Eligibility
- Inclusion Criteria:
All patients must be registered before start of treatment.
Inclusion criteria
In order to be eligible to participate in this study, a patient must meet all of the following criteria:- CAD diagnosis defined by the combination of:
- Chronic hemolysis (>3 months; suppressed haptoglobin) and
- Cold agglutinin titer of 64 or higher at 4°C and
- Positive direct antiglobulin test, strongly positive (at least 2+) with anti-C3d and negative or weakly positive (maximum 2+) with anti-IgG,
AND:
- The presence of a clonal B-cell lymphoproliferative disorder defined by:
- M-protein by serum electrophoresis confirmed by immunofixation, and/or
- A clonal CD20 positive lymphocyte population in the bone marrow (a very small clone visible only by flow cytometry is allowed)
- Indication for therapy:
- Hb ≤ 10.5 g/dL
AND/OR
- Considerable CIPS (grade 2 or more; see appendix E)
- Relapsed or refractory after at least one prior CAD treatment, OR is treatment naïve and not eligible for currently available treatment options, per clinician’s judgement.
- Age ≥ 18 years.
- ECOG/WHO performance status ≤ 2 (see appendix B). WHO performance status 3 is allowed if related to the CAD.
- Adequate bone marrow function as defined by:
- Absolute neutrophil count (ANC) > 1.0 × 10^9/L, and
- Platelets ≥ 100 x10^9/L.
- Ferritin levels above the lower limit of normal (LLN). Concurrent treatment with iron supplementation is permitted if the patient has been on a stable dose during the previous 4 weeks.
- Total bilirubin level above the upper limit of normal (ULN), as a measurable parameter for hemolysis, and that is not attributable to Gilbert's syndrome.
- Negative pregnancy test at study entry for woman of childbearing potential.* * Women of childbearing potential must be: (1) surgically sterile or ≥ 2 years after the onset of menopause; (2) willing to use a highly effective contraceptive method such as oral contraceptives, intrauterine device, or sexual abstinence during study treatment and for ≥ 90 days after last dose of zanubrutinib.
- Male patients with a female partner of childbearing potential are eligible if vasectomized or if they agree to the use of barrier contraception with other methods as described above during the study treatment period and for ≥ 90 days after the last dose of zanubrutinib.
- Written informed consent.
- Patient is capable of giving informed consent and can understand and agrees to comply with the requirements of the study and the schedule.
- CAD diagnosis defined by the combination of:
- Exclusion Criteria:
A patient who meets any of the following criteria cannot be included in this study:
- A diagnosis of agressive lymphoma, chronic lymphocytic leukemia (CLL), or the presence of overt extramedullary B-cell lymphoproliferative disease. (Note: presence of a B-cell lymphoproliferative disorder, limited to the bone marrow, including WM, MZL or IgM MGUS, is allowed. However, the indication for treatment should be only cold agglutinin-related, as per inclusion criteria.)
- Cold agglutinin syndrome (CAS) secondary to specific infection (Mycoplasma or Epstein-Barr virus) or rheumatological disorders
- Mixed AIHA, Evans syndrome (concurrent autoimmune thrombocytopenia/ITP).
- Prior non-lymphatic malignant disease within the past 3 years, except for curatively treated basal or squamous cell skin cancer, non-muscle-invasive bladder cancer, carcinoma in situ of the cervix or breast, or localized Gleason score 6 prostate cancer.
- History of severe bleeding disorder such as hemophilia A, hemophilia B, von Willebrand disease, or history of unexplained spontaneous bleeding requiring blood transfusion or other medical intervention.
- History of stroke or intracranial hemorrhage within 6 months before first dose of study drug.
- Previous treatment with BTKi.
- Major surgery within 4 weeks of the first dose of study drug.
- Requiring ongoing treatment with warfarin or warfarin derivatives. Please note: Patients being treated with DOACs (e.g., apixaban, edoxaban or rivaroxaban) or antiplatelet therapy can be included, but must be properly informed about the increased risk of hemorrhage under treatment with zanubrutinib.
- Requiring ongoing treatment with a moderate or strong CYP3A inducer. Patients requiring ongoing treatment with a CYP3A inhibitor (see Appendix G) can be included (with an adjusted dose of zanubrutinib).
- Requiring ongoing treatment with systemic corticosteroids for an indication other than AIHA/CAD at a dose of > 10 mg prednisone per day.
- Previous CAD treatment within the following time frames:
- rituximab, or bortezomib monotherapy within 3 months prior to inclusion
- bendamustine, fludarabine or other cytotoxic therapy with or without rituximab, or bortezomib with rituximab, within 4 months prior to inclusion
- anticomplement therapies within 5 half-lives of the specific drug prior to inclusion
- Erythropoietin use, which has not been at a stable dose within 3 months prior to inclusion
- Clinically significant cardiovascular disease including one of the following:
- Myocardial infarction within 6 months before screening
- Unstable angina within 3 months before screening
- New York Heart Association (NYHA) class III or IV congestive heart failure (see appendix D)
- History of clinically significant arrhythmias (eg, sustained ventricular tachycardia, ventricular fibrillation, torsades de pointes)
- QTcF > 480ms based on Fridericia’s formula
- History of Mobitz II second-degree or third-degree heart block without a permanent pacemaker in place
- Uncontrolled hypertension as indicated by a minimum of 2 consecutive blood pressure measurements showing systolic blood pressure > 170 mmHg and diastolic blood pressure > 105 mmHg at screening
- Severe or debilitating pulmonary disease (CTCAE grade III-IV, see appendix D).
- Unable to swallow capsules or disease significantly affecting gastrointestinal function such as malabsorption syndrome, resection of the stomach or small bowel, bariatric surgery procedures, symptomatic inflammatory bowel disease, or partial or complete bowel obstruction.
- Significant renal dysfunction (creatinine clearance < 30 mL/min after rehydration as estimated by the Cockcroft-Gault equation or the Modification of Diet in Renal Disease [MDRD] equation or as measured by nuclear medicine scan or 24-hour urine collection).
- Significant hepatic dysfunction (transaminases ≥ 2.5 times ULN and/or serum direct bilirubin ≥ 2 x ULN). ( Elevated indirect (unconjugated) bilirubin is accepted if this is related to active hemolysis.
- Uncontrolled HIV infection. Patients with HIV positivity but controlled infection (= sustained negative viral load) may be enrolled.
- Known hepatitis B infection with serologic status: presence of hepatitis B surface antigen (HBsAg) or hepatitis B core antibody (HBcAb). Patients with presence of HBcAb, but absence of HBsAg, are eligible if HBV DNA is undetectable (< 20 IU/mL), and if they are willing to undergo monitoring for HBV reactivation.
- Presence of Hepatitis C (HCV) antibody.
- Active fungal, bacterial and/or viral infection requiring systemic therapy.
- Known active or latent mycobacterial infection.
- Vaccination with a live vaccine within 28 days prior to the first dose of study drug.
- Pregnant or breastfeeding women.
- Current participation in another interventional clinical trial.
- Underlying medical conditions that, in the investigator’s opinion, will render the administration of study drug hazardous or obscure the interpretation of toxicity or AEs, including severe neurological, psychological, familial, sociological or geographical condition which would compromise ability to comply with study procedures, or is accompanied by a life expectancy of < 6 months.
- History of intolerance to the active ingredients or other ingredients of zanubrutinib.
Registration Details
Eligible patients should be registered before start of treatment. Patients need to be registered at HOVON by one of the following options:
• HOVON registration database (see HOVON website www.hovon.nl for the most recent link to ALEA). Account for registration and a registration manual can be requested at HOVON.
• By sending the completed registration CRF by e-mail to hovon@erasmusmc.nl Monday through Friday, from 09:00 to 17:00 CET.
• By phone +31.10.7041560 Monday through Friday, from 09:00 to 17:00 CET
The following information will be requested at registration:
• Protocol number
• Institution name
• Name of caller/responsible investigator
• Sex
• Year of birth
• Age
• Date written informed consent
• Specific items patient gives consent for (see ICF)
• Eligibility criteria
All eligibility criteria will be checked with a checklist.
Each patient will be given a unique patient study number (a sequence number by order of enrollment in the trial). Patient study number will be given immediately by the online registration database or by phone and confirmed by email.
Participating Sites
Ziekenhuizen die deelnemen aan het onderzoek staan benoemd op de HOVON website bij het onderzoek. Het kan zijn dat uw ziekenhuis niet genoemd wordt, maar wel aan het onderzoek deelneemt. Informeer hiernaar bij uw arts.