GCLLSG HO500 CLL
Main info
- Identificatie:
- HO500 CLL
- Sponsor:
- GCLLSG
- Working group party:
- CLL
- Age:
- >= 18
- Stadium:
- 1st lijn
- Echelon:
- Level D
- Included patients:
-
901(of 897)
- Active sites:
-
25(of 26)1 sites are pending
- Title:
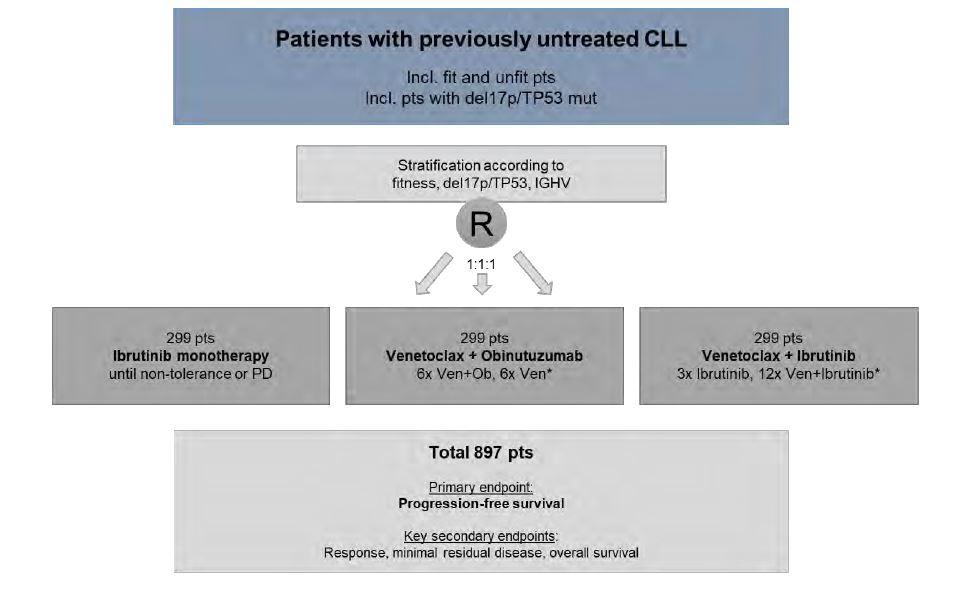
A phase 3 multicentre, randomized, prospective, open-label trial of ibrutinib monotherapy versus fixed-duration venetoclax plus obinutuzumab versus fixed-duration ibrutinib plus venetoclax in patients with previously untreated chronic lymphocytic leukaemia (CLL)
Timeline
News
21-Sep-2022: please note that the study currently uses a waiting list for the inclusion of the last patients. This waiting list is full, therefore no new patients can be enrolled / be placed on the waiting list.
27-Jul-2021: A new version of the contact details has been uploaded below under A1.
09-Jul-2021: The study has been approved in Belgium by the EC UZ Leuven on 06-Jul-2021. Please find all study documents below.
Flow
Details
- Phase:
- Prospective Phase III study
- Monitoring Type:
- Study Specific
- Objectives:
The primary objective of the study is to compare the efficacy of continuous ibrutinib monotherapy with fixed-duration venetoclax plus obinutuzumab and fixed-duration ibrutinib plus venetoclax by measuring progression-free survival (PFS) in patients with previously untreated CLL.
Eligibility
- Inclusion Criteria:
- Documented CLL/Small lymphocytic lymphoma (SLL) requiring treatment according to iwCLL criteria.
- Age at least 18 years.
- Life expectancy ≥ 6 months.
- Ability and willingness to provide written informed consent and to adhere to the study visit schedule and other protocol requirements.
- Adequate bone marrow function independent of growth factor or transfusion support within 2 weeks of screening initiation as follows, unless cytopenia is due to CLL:
- Absolute neutrophil count ≥ 1.0 × 109/L
- Platelet counts ≥ 30 × 109/L; in cases of thrombocytopenia clearly due to CLL (per the discretion of the investigator), platelet count should be ≥ 10 × 109/L
- Total haemoglobin ≥ 8 g/dL (without transfusion support, unless anaemia is due to CLL)
- GFR >30ml/min directly measured with 24hr urine collection, calculated according to the modified formula of Cockcroft and Gault (for men: GFR ≈ ((140– age) x bodyweight)/ (72 x creatinine), for women x 0, 85) or an equally accurate method.
- For patients with creatinine values within the normal range the calculation of the clearance is not necessary. Dehydrated patients with an estimated creatinine clearance less than 30ml/min may be eligible if a repeat estimate after adequate hydration is > 30 ml/min.
- Adequate liver function as indicated by a total bilirubin ≤ 2 x, AST/ALT ≤ 2.5 x the institutional ULN value, unless directly attributable to the patient’s CLL or to Gilbert’s Syndrome.
- Negative serological testing for hepatitis B (HbsAg negative and anti-HBc negative; patients positive for anti-HBc may be included if PCR for HBV DNA is negative and HBV-DNA PCR is performed every month until 12 months after last treatment cycle), and for hepatitis C (anti-HCV-ab negative; in case of positive HCV anti-body test, negative HCV-PCR is required).
- Eastern Cooperative Oncology Group Performance Status (ECOG) performance status 0-2.
- Exclusion Criteria:
- Any prior CLL-specific therapies (except corticosteroid treatment administered due to necessary immediate intervention; within the last 10 days before start of study treatment, only dose equivalents up to 20 mg prednisolone are permitted).
- Transformation of CLL (Richter transformation). When Richter transformation is suspected, PET-CT and/or biopsy should be performed to rule out transformation.
- Patients with a history of PML.
- An individual organ/ system impairment score of 4 as assessed by the CIRS definition limiting the ability to receive the study treatment or any other life-threatening illness, medical condition or organ system dysfunction that, in the investigator´s opinion, could compromise the patients’ safety or interfere with the absorption or metabolism of the study drugs (e.g. inability to swallow tablets or impaired resorption in the gastrointestinal tract).
- Malignancies other than CLL currently requiring systemic therapies, not being treated with curative intent before (unless the malignant disease is in a stable remission due to the discretion of the treating physician or showing signs of progression after curative treatment.
- Uncontrolled or active infection.
- Patients with known infection with human immunodeficiency virus (HIV).
- Requirement of therapy with strong CYP3A4 and CYP3A5 inhibitors/inducers (incl. up to 7 days prior to study treatment start).
- Anticoagulant therapy with warfarin or phenprocoumon, (alternative anticoagulation is allowed (e.g. DOACs), but patients must be properly informed about the potential risk of bleeding under treatment with ibrutinib).
- History of stroke or intracranial hemorrhage within 6 months prior to registration for study screening.
- Known bleeding disorders
- Child B / C liver cirrhosis
- Use of investigational agents which might interfere with the study drug within 28 days prior to registration for study screening.
- Vaccination with live vaccines 28 days prior to registration for study screening.
- Major surgery less than 30 days before start of study treatment.
- History of severe allergic or anaphylactic reactions to humanized or murine monoclonal antibodies, known sensitivity or allergy to murine products.
- Known hypersensitivity to any active substance or to any of the excipients of one of the drugs used in the trial.
- Pregnant women and nursing mothers (a negative pregnancy test is required for all women of childbearing potential within 7 days before start of study treatment; further pregnancy testing will be performed monthly).
- Fertile men or women of childbearing potential unless:
- surgically sterile or ≥ 2 years after the onset of menopause
- willing to use two methods of reliable contraception including one highly effective contraceptive method (Pearl Index <1) and one additional effective (barrier) method during study treatment and for 18 months after the end of study treatment.
- Legal incapacity.
- Prisoners or subjects who are institutionalized by regulatory or court order.
- Persons who are in dependence to the sponsor or an investigator.
Registration Details
Eligible patients should be registered before start of treatment.
A central medical review of the screening eCRF and the results of the screening assessments in the central laboratories will be performed by GCLLSG study physicians for verification of the eligibility of the patient, especially for confirmation of previously untreated CLL. Approval of enrolment by the GCLLSG central study office is mandatory before randomization and initiation of study treatment. Additionally, the GCLLSG study office will notify the sites if a patient is potentially at increased risk for development of TLS based on the baseline assessments.
At enrolment approval all the tests/ assessments relevant for the screening process should not be older than 42 days (exception: CT/ MRI scan results are acceptable within a time frame of 56 days before enrolment approval or other timeframe agreed with GCLLSG study office). Patients will be randomly assigned to treatment groups through 1:1:1 randomization process with stratification according to del(17p)/ TP53, IGHV and fitness (CIRS with a cut-off of 6 points and/ or GFR <70 ml/min). Treatment has to start within 14 days after randomization, but not later than 28 days after enrollment approval, i.e. approval of the patient by the GCLLSG study office.